Congenital Anomalies
Tatyana Milman
Introduction
Congenital anomalies are defects present at birth. The frequency of congenital ocular malformations has been estimated to be up to 6.8 per 10,000 live births.1,2 Malformations of the eye and its surrounding tissues can occur in isolation, or as a part of a broader syndrome. Ocular malformations can have genetic and nongenetic causes. Those with genetic etiology can be familial or sporadic, and the responsible chromosome or gene may have been identified. Nongenetic causes include infectious agents, teratogens, and disruption or deformation of ocular structures by intrauterine environment (e.g., amniotic band syndrome or oligohydramnios). Often, however, the specific cause of a given malformation is unknown.3,4,5
This chapter provides an overview of congenital malformations of the eye and the ocular adnexa, with an emphasis on pathology. In-depth reviews of embryology and molecular and developmental mechanisms that guide ocular development of disorders discussed in this chapter can be found elsewhere in the text.
Developmental Anomalies
In this section, the ocular malformations are approached on an anatomic basis. The disorders that affect the entire eye, the orbit and ocular adnexa, anterior and posterior segments of the eye, and selected developmental syndromes are discussed. Developmental biology of the eye is briefly summarized to facilitate the understanding of developmental anomalies described.
Disorders that Affect the Entire Eye
Developmental biology
Optic primordium becomes first recognizable at approximately 22 days of gestation, appearing as optic sulci, or bilateral grooves, in the neural fold of the embryo. The optic pits develop from continued evagination of the sulci and eventually form optic vesicles at approximately 25 days of gestation. The optic vesicles remain attached to the neural tube by optic stalks and become surrounded by neural crest cells. As the optic vesicles grow laterally and approach the surface ectoderm, the overlying ectodermal cells are induced to thicken and form the lens placode at 28 days of gestation. The lens placode will later invaginate to form a lens vesicle and, ultimately, the lens nucleus. Simultaneously with the development of the lens, differential growth and movement of the cells of the optic vesicle result in the invagination of its temporal and inferior walls, thus forming an optic cup. The outer layer of the optic cup will evolve as the iris pigment epithelium, iris sphincter and dilator muscles, ciliary body pigmented epithelium, and retinal pigmented epithelium. The inner layer of the optic cup will eventually form iris pigment epithelium, nonpigmented ciliary body epithelium, and the retina. At first, the two layers of the developing cup have a small space between them, which is obliterated as the inner layer becomes juxtaposed to the outer layer. Initially, the cup is incomplete at its inferior portion with a gap between the margins of the cup. This embryonic fissure (also known as choroidal, or fetal, or optic fissure) transmits the hyaloid vascular system beginning at approximately 29 days of gestation. The two lips of the optic fissure meet and initially fuse at the midregion of the cup near the equator of the globe. The closure then proceeds anteriorly to the rim of the cup and posteriorly down the stalk, enclosing the hyaloid artery, and is completed by approximately 33 days of gestation. Morphogenesis of the optic cup influences the development of surrounding tissues. The surrounding neural crest contributes to formation of the corneal stroma and endothelium, trabecular meshwork, iris and ciliary body stroma, ciliary body muscle, choroid, sclera, the orbit, and adnexa.3,5,6
Classification of anomalies in organogenesis and selected conditions
Deformities affecting the entire eye have their origin at the early stages of embryonic life before the closure of the embryonic fissure has completed the formation of the optic cup. These anomalies can be classified into the following categories7:
Abnormalities occurring in association with the development of the primary optic vesicle
Anophthalmos
Anophthalmos refers to the total absence of the ocular tissue in the orbit. It is a rare congenital malformation, with an estimated frequency of 4.18 in 100,000 births.2 In most cases, however, a small rudimentary eye (extreme microphthalmos) could be present that may be clinically indistinguishable from true anophthalmos. The diagnosis of true anophthalmos, therefore, can be made only after serial sectioning and histopathologic examination of the orbital tissues.8 Three types of the anophthalmos are recognized.
Primary anophthalmos: caused by the suppression of formation of the optic vesicle before the 2-mm stage of the embryogenesis.9 The orbit contains no neuroectodermally derived structures, nor any structures dependent on the optic cup for development.10
Secondary anophthalmos: results from the complete suppression or grossly anomalous development of the entire anterior portion of the neural tube and is incompatible with life.11
Consecutive or degenerative anophthalmos: caused by initial formation, but Later, regression of the optic vesicle.3 Histopathologically, no neuroectoderm is present, but ocular structures are present whose embryologic development is dependent on the initial presence of the optic vesicle.
Primary anophthalmos can be isolated or associated with a broader syndrome. When isolated, it is usually bilateral and may have autosomal recessive inheritance.12 Primary anophthalmos has been associated with several chromosomal anomalies (e.g., trisomy 13 and microdeletion syndromes. Recently, mutations in SOX2 gene located on chromosome 3q have been implicated in some cases of isolated and syndromic anophthalmos.13,14,15
Cyclopia and Synophthalmos
Cyclopia and synophthalmos are lethal conditions that occur with a frequency of approximately 1 in 13,000 to 1 in 20,000 live births. Cyclopia is characterized by the presence of a single eye or an ocular structure in the upper median portion of the face. The orbital, nasal, and oral regions are deformed, displaying a single median orbit, fusion of maxillary processes with absence of the nasal bones and passages, hypoplastic oral cavity, and general midline hypoplasia. The nose is usually present as a rudimentary proboscis above the pseudo-orbit, but may occasionally be absent. Synophthalmos is a much more common form, in which two incomplete globes are present in differing degrees of fusion.16,17,18
Cyclopia and synophthalmos are conditions in which the face predicts the brain. They are associated with holoprosencephaly, characterized by midline abnormalities, including “fusion” of the normal, bilateral cerebral hemispheres into a single hemisphere. It is believed that loss of inhibitory influence of precordal mesoderm on midline neural development is responsible for anomalous development of the anterior brain and for creation of a midline eye structure. Midline expression of PAX6 and ET genes in experimental models, as well as loss of function mutations in sonic hedgehog gene, have been associated with synophthalmia or cyclops and holoprosencephaly.19,20,21 In addition, chromosomal studies may reveal trisomy 13, rarely 13q-, and 18p- karyotypes.16
Histopathologically, the single eye or two partially fused eyes may display all ranges of pathology, from relatively normal to completely anomalous. The fusion of the eyes in synophthalmos is always more marked posteriorly.17,18,22 The degree of disorganization within the ocular structure tends to be greater medially. Cases that have an associated chromosomal anomaly, such as trisomy 13, are usually more disorganized and may display heterotopic elements, such as cartilage (Fig. 2.1).23
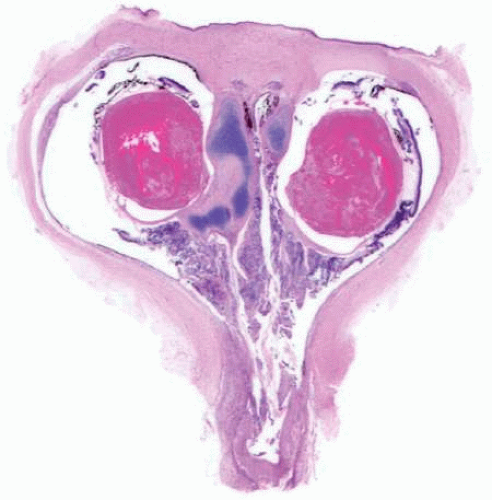 Figure 2.1. Synophthalmos in a child with trisomy 13 displays partial fusion of the two globes, which is more marked posteriorly. Intraocular cartilage, associated with ciliary body coloboma, is present medially, and is surrounded by dysplastic retina (stain, hematoxylineosin [H&E]). (Courtesy of Ralph C. Eagle, Jr., MD.) |
Abnormalities occurring after the development of the primary optic vesicle, affecting its invagination to form the optic cup:
Aberrations associated with the involution of the primary optic vesicle (e.g., congenital cystic eye, congenital nonattachment of the retina)
Aberrations associated with the closure of the embryonic cleft (e.g., coloboma)
Congenital Cystic Eye
Congenital cystic eye is a rare congenital anomaly, which occurs because of an arrest of invagination of the primary optic vesicle between the 2-mm and 7-mm stages of fetal development.7,24 Clinically, a cystic structure is observed in an anophthalmic-appearing orbit (Fig. 2.2 A). Ipsilateral eyelids may display various anomalies, such as dermal appendages, accessory limbs, and skin tags. The condition can be unilateral, bilateral, or associated with a microphthalmos with cyst and eyelid colobomas in the fellow eye. It can occur in isolation or be a part of a broader syndrome. The etiology of congenital cystic eye remains obscure, but no hereditary tendency has been established.25,26,27,28
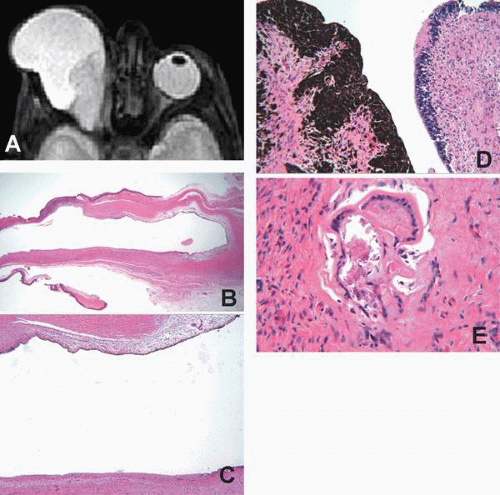 Figure 2.2. A: Axial T2W magnetic resonance imaging (MRI) scan reveals a variably enhancing mass in the right orbit. The lesion is composed of anterior cystic and posterior solid components. No recognizable ocular structures are observed. The left eye appears radiographically normal. (Courtesy of Edward Howes, MD B-D. Presented by Edward Howes, MD at Eastern Ophthalmic Pathology Society meeting 2005.) B: Low magnification photomicrograph highlights the cystic nature of the lesion. Higher magnification photomicrographs (C,D) reveal that the cyst is lined by neuroglial, ependymal, and primitive ciliary body and choroidal epithelia. A focus of lens tissue with folded capsule is also observed within the neuroglial proliferation (E) (stain, hematoxylin-eosin [H&E]). |
Histopathologically, a unilocular or multilocular cyst lined by neuroglial tissue is observed. Occasionally, the lesion may be solid and filled completely with neuroglial tissue. No recognizable ocular structures are present if the arrest of development of the optic vesicle occurred at an early stage.25,26,27,28 If the insult occurred later in development, malformed and incompletely developed lens, cornea, iris, ciliary body, choroid, and retina may be present (Fig. 2.2 B–E).28,29,30,31 Malformed optic stalk lined by ependymal tissue has been described, but often an optic nervelike structure is not observed.26,27
Congenital Nonattachment of the Retina
It has been suggested that persistent nonattachment of the retina occurs because of aberrant differential growth and, consequently, impaired juxtaposition of the two layers of the optic cup.32 This condition has been observed unilaterally and bilaterally, in microphthalmic and normally sized eyes, as an isolated anomaly or as a part of a broader syndrome. Shallow anterior chamber, retrolental white mass, cataract, no light perception vision, and nystagmus are typical clinical findings.33,34,35 Sporadic and familial (mostly autosomal recessive) forms have been described. Recent studies by Ghiasvand et al.35,36 have mapped the retinal nonattachment gene to chromosome 10.
Histopathology of congenital nonattachment of the retina is relatively nonspecific. The retina is detached and may contain dysplastic rosettes and tubules. Persistent hyperplastic primary vitreous, glial proliferations, and, less often, vascular proliferations and hemorrhage are present. Some of these findings may be encountered in trisomy 13, persistent hyperplastic primary vitreous, Norrie’s disease, and multiple other conditions associated with retinal detachment in infancy.33,34 Clinical examination and history, therefore, are most helpful in establishing the correct diagnosis.
Coloboma
Typical colobomas occur as a result of defective closure of the embryonic fissure at the 7-mm to 20-mm stage of development.7,32 They are observed inferonasaly at any point along the course of the embryonic fissure, and may involve the iris, ciliary body, retina or choroid, and the optic nerve. Individual structure, all structures, or any combination of the above may be involved (Fig. 2.3 A).
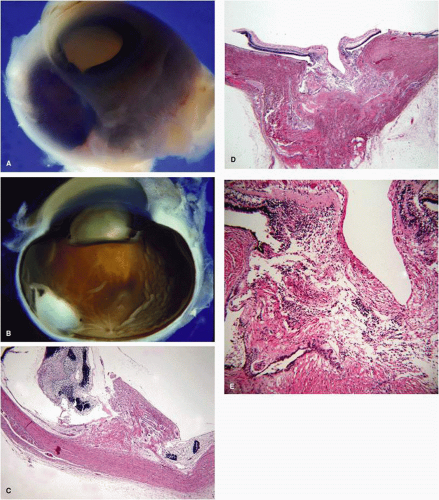 Figure 2.3. A: Transillumination of the globe highlights the colobomatous defects in the iris and posterior choroid. In this eye, the arrest of closure of embryonic fissure has occurred shortly after the initial fusion of the lips of the optic cup at the equator of the globe. B: Retinochoroidal coloboma manifests the defects in the retina, choroid, and the retinal pigment epithelium (RPE). The underlying sclera is thin and slightly bowed posteriorly. (Courtesy of Ralph C. Eagle, Jr., MD.) C: Histopathologically, retinochoroidal coloboma shows lack of choroid and normal retina within the colobomatous defect. Dysplastic retina is observed at the edges of the defect, and the defect itself is filled by a gliotic tissue. D,E: Photomicrograph of the coloboma of the optic nerve displays absence of peripapillary choroid, RPE, and normal retina. Neuroglial tissue prolapses into a colobomatous defect, lined by an ectatic sclera (stain, hematoxylin-eosin [H&E]). |
Isolated colobomas can be inherited as an autosomal dominant disorder, although autosomal recessive inheritance also occurs.37 They may be associated with other ocular anomalies (microcornea, microphthalmos)38 and broader syndromes (coloboma; Hart anomaly; atresia, choanal; retardation of mental and somatic development; genital anomalies; ear anomalies [CHARGE]),39 Patau syndrome (trisomy 13), and cat eye syndrome (iris coloboma, anal atresia, facial dysmorphism; locus on chromosome 22).40
Histopathologically16:
The iris coloboma shows a complete absence of all tissue in the involved area.
The ciliary body coloboma shows the defect filled with mesodermal and vascular tissues and, even cartilage, in trisomy 13. Hyperplastic ciliary processes are observed at the edges of the coloboma.
The choroidal coloboma shows an absence or atrophy of choroid and an absence of the retinal pigment epithelium (RPE). The RPE is hyperplastic at the edges of the defect. The overlying retina is atrophic, gliotic, and may contain dysplastic rosettes. The sclera in the region of the defect is usually thinned and may be cystic. The cystic space often is filled with glial tissue (Fig. 2.3 B,C).
The optic nerve coloboma shows a large defect at the side of the nerve involving the retina, RPE, and choroid. Atrophic and gliotic retina lines the defect. The sclera is ectatic and bowed posteriorly. The wall of the defect may contain adipose tissue and even smooth muscle cells (Fig. 2.3 D,E).
It is important to note that the primary defect in a retinochoroidal coloboma lies in the absence of neuroectoderm, and the neural crest derived tissues, such as iris stroma, ciliary body stroma and muscle, and the choroid are absent secondarily.
MICROPHTHALMOS WITH CYST is a special type of coloboma caused by an incomplete closure of the embryonic fissure between 7-mm and 14-mm stages of fetal development.7,24 In this condition, the contents of a microphthalmic eye communicate through a colobomatous defect with a cystic cavity, lined by an ectatic sclera and neuroectodermal and glial tissues (Fig. 2.4 and 2.5).16 It is believed that failure of closure of the embryonic fissure prevents the normal establishment of intraocular pressure necessary for globe expansion, thus resulting in a microphthalmos. Although microphthalmos with cyst is more commonly an isolated unilateral anomaly, it can be bilateral and associated with other congenital malformations.41 Most cases are sporadic, but some may be familial (likely autosomal recessive),42 and some are associated with chromosomal anomalies (13q deletion, partial monosomy 18, translocation t[3;5]).16,43
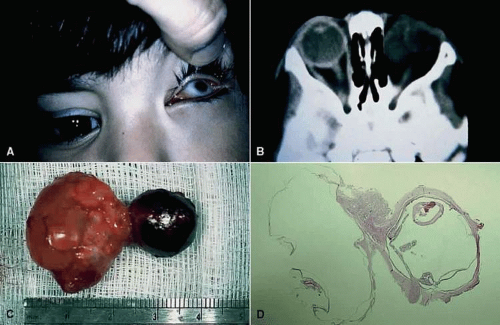 Figure 2.4. Microphthalmia with cyst. A: The microphthalmic left eye in a 2-year-old girl. B: Computed tomographic scan showing the cyst nasal to the microphthalmic eye. C: Gross specimen, showing that the cyst is larger than the eye. D: Low-power photomicrograph showing the relatively well-formed eye and large cyst (stain, hematoxylin-eosin [H&E] × 1). There is focal retinal dysplasia. (UTHSC-SA EP 169. Courtesy of Charles R. Leone, MD) |
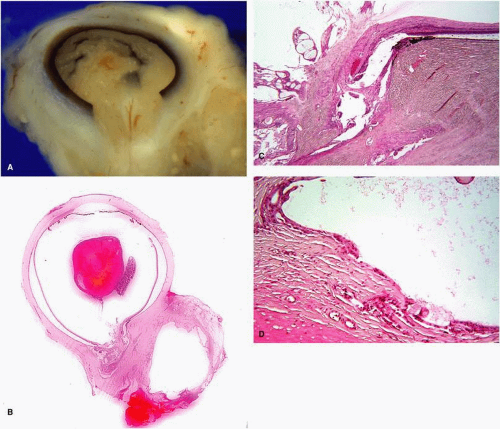 Figure 2.5. Another example of a microphthalmos with cyst. A: Disorganized contents of a microphthalmic eye prolapse through a large defect in its wall into a cystic cavity lined by ectatic sclera. B: Low power photomicrograph illustrates communication of a microphthalmic globe with a cystic cavity. C: Disorganized intraocular tissue prolapses into a large colobomatous defect. Note the termination of the choroid and normal retina at the edges of the defect. D: The cyst is lined by neuroglial tissue and ectatic sclera (stain, hematoxylin-eosin [H&E]). |
Abnormalities resulting from a failure in development of the eye as a whole
Microphthalmos
Microphthalmos is defined as the eye with an axial length less than 15 mm in greatest diameter at birth and less then 18.5 mm in adults.7,16 This malformation occurs with a frequency of approximately 1.5 per 10,000 live births.44 Two types of microphthalmos are recognized:
Simple microphthalmos or nanophthalmos:
In this condition, the eye is smaller than normal size but has no other gross abnormalities. Typically, the lens is disproportionably large for the size of the globe, and the posterior chamber is disproportionably small for the size of the anterior segment.45 The sclera of nanophthalmic eyes is thickened and may contain glycosaminogycanlike deposits between irregularly arranged and variably sized collagen bundles. These abnormalities in nanophthalmos sclera may contribute to its inelasticity, thereby impeding the blood flow through the vortex veins, and predisposing the eye to uveal effusion.46,47
Simple microphthalmos can occur as an isolated anomaly, but can also be associated with other systemic disorders, such as fetal alcohol syndrome, hypotonic dystrophy, and achondroplasia, which may implicate decreased size of the optic cup, altered vitreous proteoglycans, low intraocular pressure, and abnormal release of growth factors in the pathogenesis of microphthalmos.45
Complex microphthalmos:
In this condition, the eye is not only small, but also has other anomalies, such as retinochoroidal coloboma, microcornea, anterior segment dysgenesis, cataract, persistent hyperplastic primary vitreous, and retinal dysplasia.48
Complex microphthalmos is associated with a multitude of syndromes, including CHARGE, Aicardi syndrome, Lenz microphthalmia syndrome, oculodentodigital dysplasia, and many others.49,50,51 In addition, congenital infections (rubella, parvovirus, herpes simplex virus type 2, toxoplasmosis, and cytomegalovirus [CMV]) and teratogens (alcohol, retinoic acid, methotrexate, and vincristine) may be associated with complex microphthalmos.49
Sporadic, autosomal dominant, autosomal recessive, and X-linked modes of inheritance have been observed, and numerous chromosomal abnormalities have been associated with this condition. It is believed that any disorder that affects telencephalon and cranial neural crest development can result in microphthalmos.51,52,53 In addition, Weiss et al.48 suggested that inadequate production of secondary vitreous may result in complex microphthalmos.
Disorders that Affect the Orbit, Eyelids, and Adnexa
Developmental Biology
The bones, cartilage, fat, and connective tissues of the orbit arise from the frontonasal and maxillary processes of undifferentiated neural crest cells that surround the optic cups by the fourth week of gestation.5 All bones of the orbit form through endomembranous ossification, except for the ethmoid and the temporal part of the sphenoid, which undergo endochondral ossification.54 Ossification begins in the fifth month of gestation, and fusion of the bones occurs between the sixth and seventh months of gestation.5
The extraocular muscles arise from myogenic paraxial mesoderm. Their development is influenced by the interactions with the neural crest mesenchyme.55 Myoblasts with myofibrils and immature Z bands are apparent by the fifth week of gestation. The tendons of the extraocular muscles fuse with the sclera in the third month of gestation.56,57
The eyelids initially develop as a proliferation of the surface ectoderm at the end of the first month of gestation, and become recognizable as skin folds that surround mesenchyme of neural crest origin. Later, mesodermal mesenchymal cells infiltrate the eyelid and differentiate into the eyelid muscles and vasculature. The eyelid folds grow laterally and toward each other, fusing together approximately at 10 weeks of gestation. The cilia and the glands within the eyelids develop from invaginations of epithelial cells into the underlying mesenchyme, and mature while the eyelids are fused. Production of sebum from the sebaceous glands and keratinization of the epidermis contribute to the separation of the eyelids late in the fifth month of gestation.5
Abnormalities in Eyelid Development
When the normal development of the eyelids is interrupted, a wide range of congenital malformations may occur.
Cryptophthalmos (Ablepharon)
Failure of the eyelid folds to form correctly during embryonic development results in cryptophthalmos. Three varieties of cryptophthalmos have been described:
In complete cryptophthalmos, the globe is covered by a continuous layer of skin, which extends uninterrupted from the forehead to the cheek. Occasionally, linear dimpling of the skin may be present, representing an aborted eyelid fold.
In incomplete cryptophthalmos, the medial eyelids are not developed and fused, but the lateral structures are intact.
Cryptophthalmos of symblepharon variety presents with fusion of the eyelid skin to the globe.
The underlying globe is malformed. Although the lens and posterior pole are usually normal, the cornea and anterior segment structures are typically absent or mal developed (e.g., Peters anomaly). The globe may be microphthalmic (Fig. 2.6).58,59
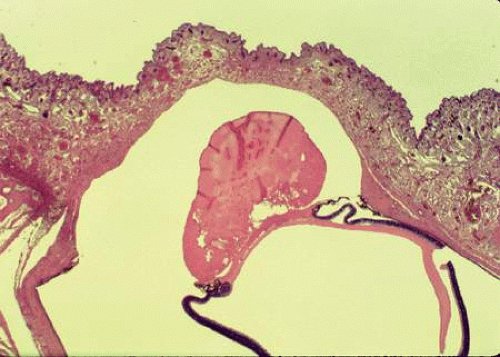 Figure 2.6. An eye with complete cryptophthalmos demonstrates lack of development of normal eyelid structures. The globe is covered by a strip of skin. Numerous abnormalities in the underlying anterior segment structures are present, including absence of the cornea and the iris and malformation of the anterior chamber angle and the lens (stain, hematoxylin-eosin [H&E]). (Courtesy of Ralph C. Eagle, Jr. MD.) |
Cryptophthalmos can be bilateral or unilateral, isolated or syndromic. Sporadic, autosomal dominant, and autosomal recessive modes of inheritance have been described. The most common syndromic cause of cryptophthalmos is Fraser syndrome, which has an autosomal recessive inheritance. In Fraser syndrome, cryptophthalmos is accompanied by facial dysmorphism, syndactyly, urogenital anomalies, and cognitive impairment. Some, but not all, patients with Fraser syndrome may have loss of function mutations in the extracellular matrix protein FRAS1, expressed in the basement membranes of skin, corneal, kidney, and gut epithelia. The FRAS1 gene is located on chromosome 4.58,60,61
Eyelid Coloboma
An eyelid coloboma is a congenital full thickness defect of the eyelid involving the eyelid margin, with accompanying absence of the eyelashes and glands.62 The defect can range from notching at the eyelid margin to complete absence of a segment of the eyelid (Fig. 2.7).16 This congenital malformation can result from a failure of the eyelid folds to meet or from premature separation of the eyelids during development. It can also occur as a result of disruption of fetal eyelid tissues, as seen in amniotic band syndrome. Eyelid colobomas can be sporadic or familial, isolated or syndromic, unilateral or bilateral.62
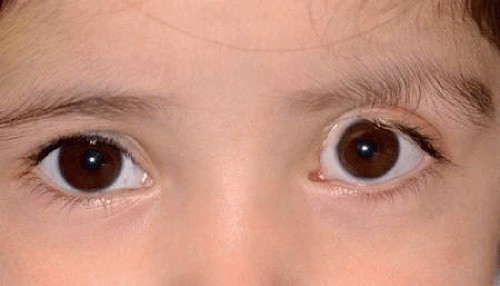 Figure 2.7. Sporadic unilateral coloboma of the left upper eyelid shows a full thickness defect in the medial eyelid. |
The most common location is in the upper eyelid, between the medial third and lateral two-thirds. Upper eyelid colobomas are associated with Goldenhar syndrome (oculoauriculovertebral dysplasia). This syndrome is characterized by eyelid colobomas, epibulbar dermoids, accessory auricular appendages, aural fistulas, deafness, hemifacial hypoplasia of the soft and bony tissues, and vertebral anomalies. Although most cases of Goldenhar syndrome are sporadic, some have autosomal dominant inheritance.16,63,64
Colobomas of the lower eyelid is usually present at the junction of the lateral third and medial two-thirds.62 They may be associated with Treacher Collins-Franceschetti syndrome (mandibulofacial dystosis), characterized by antimongoloid slant and shortening of palpebral fissures, lower eyelid colobomas, dystopia of lateral canthi, notching of eyebrows and upper eyelids, hypoplastic or absent zygomatic arches, micrognathia, microstomia, microtia, and ear anomalies. This syndrome has an autosomal dominant inheritance with variable penetrance and expressivity.65
Ankyloblepharon
Ankyloblepharon is an abnormal full thickness fusion of the eyelid margins from incomplete separation of the eyelid folds. The fusion can extend along the entire eyelid margin, but is more common medially. Unlike cryptophthalmos, the eyelid structures are developed and the underlying globe is typically normal.7,62
Ankyloblepharon filiforme adnatum is a variant of ankyloblepharon, characterized by a single or multiple bands of tissue connecting the eyelid margins at the gray line. Histopathologically, these bands of tissue consist of a vascularized central core surrounded by the stratified squamous epithelium. Occasionally, this condition may resolve spontaneously postnatally.66
Ankyloblepharon can be isolated or associated with a broader syndrome, such as AEC syndrome (ankyloblepharon, ectodermal defects, and cleft lip and palate), a rare autosomal dominant disorder caused by mutations in p63 gene. P63 is a developmentally regulated transcription factor related to p53.67,68 Other reported syndromes and associations include popliteal pterygium syndrome, CHANDS (curly hair-ankyloblepharon-nail dysplasia), hydrocephalus, meningocele and imperforate anus, syndactyly, cardiac anomalies, and Edwards syndrome (trisomy 18).66 Although the eye is typically normal in nonsyndromic ankyloblepharon, an association of ankyloblepharon filiforme adnatum with infantile glaucoma and iridogoniodysgenesis has been recently described.69
Epiblepharon
Epiblepharon is a congenital eyelid anomaly in which an additional fold of skin is running horizontally below the lower eyelid margin. It is commonly associated with the loss of the eyelid crease, inward rotation of the lashes, and secondary corneal astigmatism. This condition may occur because of lack of anchoring of superficial eyelid skin to the underlying tissues. Epiblepharon is seen more commonly in Asian population. Although epiblepharon may resolve with facial growth, some cases require surgical intervention to prevent corneal injury.70
Euryblepharon
Euryblepharon is a rounding of the eyelid at the lateral canthal angle, associated with the increase in the horizontal eyelid margin width, vertical shortening of the eyelid, and inferiorly displaced lateral canthal tendon.71,72 The lower eyelids are usually involved, but the upper eyelids also may be affected. This condition can be associated with other eyelid anomalies and may be a part of a broader syndrome, such as Blepharo-cheilo-dontic syndrome (ectropion of lower eyelids, distichiasis of upper eyelids, euryblepharon, bilaterally cleft lip or palate, oligodontia, and conical crown form).72,73
Distichiasis
Aberrant differentiation of meibomian glands of the eyelid may produce a second row of eyelashes exiting from meibomian gland orifices. This congenital anomaly is called distichiasis. Distichiasis is usually a part of lymphedema-distichiasis syndrome, an autosomal dominant disorder with variable penetrance, mapped to the long arm of chromosome 16, FOXC2 transcription factor gene.74
Congenital Entropion
Congenital entropion is an eyelid anomaly characterized by inward rotation of the eyelid margin. It can occur owing to hypertrophy of the orbicularis muscle, absence of or abnormalities in the tarsal plate (e.g., tarsal hypoplasia, congenital tarsal “kinking”), or defects in lower eyelid retractors.16,62 Congenital entropion is occasionally associated with epiblepharon. This condition can be sporadic or inherited (commonly as an autosomal dominant trait), isolated or a part of a broader syndrome (trisomy 13, Duane retraction syndrome).75,76,77
Congenital Ectropion
Congenital ectropion, which is characterized by eversion of the eyelid margin, can occur because of tightening of anterior lamella.78 Primary congenital ectropion is rare, and is associated with Down syndrome.79 Most cases of congenital ectropion are secondary to other conditions affecting the eyelids, such as blepharophimosis syndrome80 (see below), lymphedema-distichiasis syndrome,74 euryblepharon and blepharo-cheilo-dontic syndrome,72,73 and dermatologic ichthyotic conditions that shorten anterior lamellae.81
Epicanthus
Epicanthus is a fold of skin that extends from the side of the nose to the upper eyelid, partially obscuring the medial canthus. Four types are recognized62:
Epicanthus tarsalis: the fold extends from superior tarsal area to medial canthus, and is more prominent in the upper eyelid.
Epicanthus palpebralis (simple epicanthus): the fold is equally distributed in the upper and lower eyelids.
Epicanthus inversus: the fold arises from the lower eyelid, terminates in the upper eyelid, and is more prominent in the lower eyelid.
Epicanthus superciliaris: the fold arises from the region of the eyebrow and extends over the lacrimal sac.
Blepharophimosis-Ptosis-Epicanthus Inversus Syndrome
Blepharophimosis-ptosis-epicantus inversus syndrome (BPES) is an autosomal dominant disorder (with a de novo mutation in 50% of the cases) characterized by shortening of the horizontal and vertical palpebral fissures (blepharophimosis), congenital ptosis, and epicanthus inversus. Congenital ectropion is another associated finding. Two types of BPES have been proposed. Type I is associated with infertility in female patients, whereas type II has no associated symptoms. Mutations in FOXL2 transcription factor gene on chromosome 3 and multiple chromosomal abnormalities in that region have been found in affected people.82,83
Hamartomatous Lesions of the Eyelids
Hamartoma is defined as abnormal proliferation of tissue elements found at their proper location.
Rhabdomyomatous Mesenchymal Hamartoma of the Eyelid
Hamartomatous lesions of the eyelids present as elevated, firm, fleshy, benign skin lesions. They can be single or multiple, and are typically associated with other ocular anomalies, including corneal leukoma, colobomatous orbital cysts, limbal dermoids, and upper eyelid colobomas. Association with Dellerman (oculocerebrocutaneous) syndrome and Goldenhar syndrome has been reported.83
Histopathologically, these lesions exhibit normal epidermal surfaces. Dermis contains randomly arranged mature striated muscle fibers associated with normal-appearing mesenchymal elements, such as adipose tissue, collagen, and blood vessels. These hamartomas have been reported under several different names, including striated muscle hamartoma, rhabdomyomatous mesenchymal hamartoma, congenital midline hamartoma, and hamartoma of cutaneous adnexa and mesenchyme.83
Choristomatous Lesions of the Eyelids
Choristoma is defined as presence of normal tissue elements at atypical location.
Phakomatous Choristoma
Phakomatous choristoma (Zimmerman’s tumor) is a rare sporadic congenital lesion, characteristically presenting as a small, firm, rubbery nodule in the medial aspect of the lower eyelid. It is believed that the lesion is of lenticular anlage origin, and can occur when a group of epithelial cells that are differentiating into lens tissue, is trapped in the subcutaneous tissue of the lower eyelid during fetal development.84,85,86
Histopathologically, the lesion is composed of islands of epithelial cells growing irregularly in a dense fibrous tissue matrix. Epithelial component consists of cuboidal cells with abundant eosinophilic cytoplasm and centrally located nuclei resembling lens epithelial cells, and large cells resembling bladder cells of Wedl. The latter cells have been show to contain lens-specific alpha, beta, and gamma crystallin proteins.86A Thick periodic acid-Schiff (PAS)-positive basement membrane material analogous to lens capsule surrounds the epithelial islands (Fig. 2.8).84,85,86
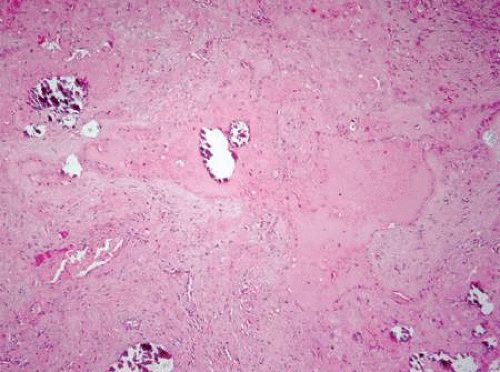 Figure 2.8. Islands of eosinophilic lens material, consisting of bladder cells of Wedl and cuboidal lens epithelial cells surrounded by a thick basement membrane (lens capsule), set in a matrix of fibrotic stroma. Foci of calcification within the lens material are also observed (stain, hematoxylin-eosin [H&E]). |
Congenital Orbital Cystic Lesions
Dermoid Cyst
Dermoid cyst is a common lesion of orbital region. It can occur as a result of sequestration of surface ectoderm “pinched off” at bony suture lines or along lines of embryonic closure. In most cases, the lesion is found superotemporally near the frontozygomatic suture, but can be found along any other suture. The patients typically present with a slowly progressive painless subcutaneous mass, which is firm to palpation, and may be attached to the underlying bone (Fig. 2.9 A). Pain and inflammatory presentation can occur if the lesion spontaneously ruptures.87,88
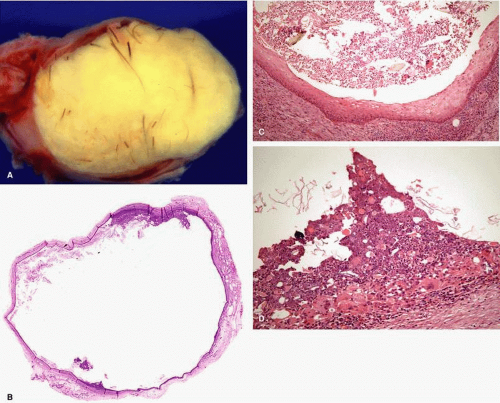 Figure 2.9. A: A cystic lesion filled with yellowish material and hair shafts was removed from the superotemporal orbit of a young man. Histopathologic evaluation reveals a cyst, lined by stratified squamous keratinized epithelium, and containing keratin and hair shafts in the lumen. No dermal appendages are present in these particular photomicrographs, but could be observed in other locations (B,C). The lesion has ruptured, inciting a severe foreign body granulomatous reaction to keratin (D) (stain, hematoxylin-eosin [H&E]). |
Histopathologically, a typical dermoid cyst is lined by a stratified squamous keratinized epithelium with dermal appendages (hair follicles, sebaceous and sweat glands). The cavity contains keratin and hair shafts (Fig. 2.9 B–D).16 Because dermoid cyst is a congenital lesion, characterized by the presence of normal tissue at atypical location, it may be regarded as a type of choristoma.16,88
A rare variant of a dermoid cyst, conjunctival dermoid cyst, has been described. Conjunctival dermoid cyst is lined by conjunctival epithelium (stratified squamous nonkeratinized epithelium with goblet cells), but contains adnexal structures in its wall and is filled with clear fluid.16,87
Other congenital orbital cysts of the surface epithelium include EPIDERMAL CYST, CONJUNCTIVAL EPITHELIAL CYST, RESPIRATORY EPITHELIAL CYST, and APOCRINE GLAND CYST. Similarly to dermoid cyst, these lesions arise from sequestration of the surface epithelium in the orbital tissues during fetal development. As their names imply, these cysts are lined by the epithelium of origin.87,88
Teratomatous Cyst (Teratoma)
Orbital teratoma is a congenital multicystic mass, composed of all three embryonic germinal cell layers (ectoderm, endoderm, and mesoderm). This sporadic condition can occur as a result of aberrant differentiation of primitive pluripotential germ cells in the orbital area. Most reported cases are unilateral, but bilateral orbital teratomas have also been described. Affected children present at birth with unilateral, often progressive, proptosis. Large lesions may be associated with facial dysmorfism.16,87
Histopathologically, teratoma is characterized by the presence of various structures, including cysts lined by surface, gastrointestinal, or respiratory epithelium. Cartilage and central nervous tissue may be observed. Some orbital teratomas may even contain formed parts of a fetus.16,87 Occasionally, the eye is disorganized and is incorporated in the teratomatous mass.89 Although orbital teratomas carry low malignant potential, several reports of malignant transformation exist in literature.90
Malformations of the Anterior Segment
At approximately 28 days of gestation, inductive signals from the optic vesicle cause thickening of the surface ectoderm and the formation of the lens placode. Invagination of the lens placode forms a hollow vesicle, which detaches from the surface epithelium at about 33 days of gestation. Lens vesicle consists of a monolayer of cells surrounded by the basement membrane, the lens capsule. Elongation of the posterior lens epithelial cells and their transformation into primary lens fibers results in formation of embryonic lens nucleus at 37 days of gestation. Migration of the mitotically active anterior epithelial cells toward the equator and their differentiation into the secondary lens fibers contribute to formation of the fetal nucleus.
Separation of the lens vesicle from the surface ectoderm marks the beginning of the development of the cornea. The surface ectoderm becomes continuous and later differentiates into corneal epithelium. Basal cells of the epithelium secrete collagen fibrils and glucosaminoglycans, which constitute primary corneal stroma. Between 5 to 6 weeks of gestation, mesenchymal (neural crest) cells migrate from the margins of the rim of the optic cup along the posterior surface of the primary corneal stroma, forming future corneal endothelium. At approximately 6 to 7 weeks of gestation another wave of neural crest cells migrates anteriorly to form keratocytes of corneal stroma, and posteriorly to contribute to formation of primary pupillary membrane and iris stroma. Descemet’s membrane first appears at the eighth week of gestation as a patchy accumulation of basement membrane material. By the third month of gestation, all corneal components are present, with the exception of Bowman’s layer. Bowman’s layer may be observed by light microscopy during the fifth month of gestation.
The sclera is formed by mesenchymal cells that condense around the optic cup. Most are neural crest derived, but some may be derived from paraxial mesoderm. The sclera initially develops anteriorly before the seventh week of gestation and gradually extends posteriorly by 12 weeks of gestation.
The anterior chamber becomes first apparent as a slitlike space between the corneal endothelium and iris pupillary membrane. By approximately 7 weeks of gestation, the anterior chamber angle is occupied by a nest of mesenchymal cells. Anterior cells of neural crest origin will develop into the trabecular meshwork. Posterior cells of mesodermal origin will develop into the channels of the iridopupillary membrane. The proposed mechanisms in the development of the anterior chamber angle and trabecular meshwork include differential growth of the vascular tunic, with consequent posterior movement of the iris and ciliary body, gradual cellular rearrangement, mesenchymal atrophy, and formation of gaplike spaces continuous with the anterior chamber angle.
The iris develops as a result of anterior growth of the optic cup. Neural crest cells and the anterior portion of tunica vasculosa lentis contribute to the development of iris stroma. The most anterior portion of the tunica vasculosa lentis becomes an iridopupillary membrane, which is later incorporated into the anterior border layer of the iris. The central portion of iridopupillary membrane atrophies near term. The vessels of posterior portion of tunica vasculosa lentis are ultimately incorporated into the iris stroma, and the pupillary portion of tunica is gradually reabsorbed beginning the third month of gestation.
The development of neuroectodermaly derived tissues of the iris lags behind the formation of the iris stroma. The iris sphincter and dilator muscles differentiate from the anterior layer of the optic cup epithelium. The iris sphincter muscle is recognized at 3 months of gestation and its development is complete by the eighth month. The dilator muscle becomes apparent after sixth month, and its development continues after birth.
Anomalies of Corneal Size, Shape, and Composition
Microcornea
Microcornea is characterized by greatest diameter less than 10 mm at birth and less than 11 mm in an adult.16,91 The condition can be isolated or occur in association with microphthalmos. Autosomal dominant inheritance has been described.16,44,45
Megalocornea
Megalocornea is characterized by nonprogressive enlargement of corneal diameter to more than 12 mm at birth and more than 13 mm in an adult in greatest dimension. The proposed mechanisms for this condition include a defect in formation of the optic cup in which the anterior tips of the cup fail to fuse, allowing more space for the developing cornea, spontaneous arrest of congenital glaucoma, exaggerated growth of the cornea, and abnormalities in collagen synthesis. It is usually a bilateral isolated anomaly. Autosomal dominant, recessive, and X-linked modes of inheritance have been described.92,93,94,95,96
When this condition occurs as an isolated anomaly, it is called simple megalocornea. In many instances, however, megalocornea is associated with enlarged anterior segment structures, and the condition is then called anterior megalophthalmos. Additional ocular findings in anterior megalophthalmos include ectopia lentis, pigment dispersion syndrome, posterior embryotoxon, and cataracts. Occasionally, anterior megalophthalmos may be a part of a broader syndrome (e.g., Marfan’s syndrome, osteogenesis imperfecta, mental retardation, dwarfism, and Down syndrome).92,93,94,95,96 Histopathologically, the cornea is usually normal except for its large size.16,97
Anomalies of Lens Size, Shape, and Composition
Congenital Aphakia
Congenital aphakia is a rare anomaly that can be subdivided into two forms:
Primary congenital aphakia occurs from lack of lens induction of the surface ectoderm and may be associated with mutations in PAX6 gene. No lens anlage has developed. This condition is commonly associated with severe ocular and systemic developmental anomalies, including severe microphthalmos, anterior segment aplasia, posterior retinochoroidal and optic disc colobomas, trisomy 18, and Waardenberg syndrome.16,98
Histopathologically, the lens is absent in primary congenital aphakia. Histopathologic findings of secondary congenital aphakia depend on the underlying etiology.16
Micropspherophakia
Microspherophakia refers to a small, spheric lens. This condition can occur in isolation and is inherited in autosomal recessive fashion, although autosomal dominant inheritance has been also described.99 Occasionally, microspherophakia may be a part of a broader syndrome, such as Weill-Marchesani syndrome (short stature; short, stubby fingers; broad hands; joint stiffness mostly autosomal recessive inheritance) and Marfan’s syndrome (tall stature, long limbs, arachnodactyly, joint laxity, aortic aneurisms, autosomal dominant inheritance).99
Pathogenesis of microspherophakia may be related to arrest in development or aberrant development of secondary lens fibers, which could occur from insufficient nutrition from the tunica vasculosa lentis. Alternatively, microspherophakia may result from abnormalities in the zonular apparatus, accounting for high frequency of lens dislocation associated with this condition.99
Anterior Lenticonus (Lentiglobus)
The anterior surface of the lens can assume an abnormal conical (lenticonus) or spheric (lentiglobus) shape. Clinically, an “oil droplet” red reflex is present. The conditions may arise because of aberrant separation of the primary lens vesicle from the surface ectoderm. Although anterior lenticonus and lentiglobus can occur as isolated anomalies, more than 90% of cases of bilateral anterior lenticonus are associated with Alport’s syndrome (hemorrhagic nephritis, deafness, anterior lenticonus, anterior polar cataract, retinal flecks, retinal and iris neovascularization, autosomal dominant with incomplete penetrance and varying expressivity).16,100
Histopathologically, thinning of the anterior lens capsule, a decrease in number of anterior epithelial cells, and bulging of the anterior cortex are observed.16 With electron microscopy, anterior capsules of the patients with Alport’s syndrome reveal ultrastructural fragility and collagen IV abnormality.101,102
Posterior Lenticonus (Lentiglobus)
Posterior lenticonus is characterized by a spheric elevation on the posterior surface of the lens. Clinically, an oil dropletlike red reflex is seen. Posterior lenticonus usually occurs as a sporadic, isolated, and unilateral anomaly in association with unilateral congenital cataract, but bilateral cases of isolated posterior lenticonus with X-linked inheritance have been described. Posterior lenticonus may be occasionally associated with other ocular abnormalities, such as microphthalmos, microcornea, persistent anterior hyaloid vasculature, and uveal colobomas.103,104,105 It may also occur as a part of Alport’s syndrome106 and oculocerebrorenal syndrome of Lowe (systemic acidosis, renal rickets, hypotonia, congenital cataracts, X-linked recessive inheritance).107
Lens Coloboma
Lens coloboma is characterized by a notch in the lens, typically in inferonasal location. This anomaly likely occurs secondary to the focal absence, or mal development, of zonular fibers from the colobomatous ciliary body. It may occasionally occur in association with Marfan’s syndrome, where it may represent an incomplete form of ectopia lentis.108
Congenital Cataracts
Congenital cataracts occur with a frequency of 1 to 6 per 10, 000 live births. They can be associated with intrauterine infections, metabolic disorders, chromosomal abnormalities, and various syndromes, or may be inherited as an isolated ocular anomaly.
Most inherited isolated cataracts are bilateral and show autosomal dominant inheritance, but X-linked and autosomal recessive forms are also seen. Isolated familial cataracts have been mapped to loci on chromosomes 1p, 1q, 13q, 14q, 16q, 17p, 17q, and 22q.109,110
Numerous types of congenital cataracts have been identified. These include:
Total cataracts involve the whole lens and may be associated with posterior lenticonus.
Congenital morgagnian cataracts are rare, and are characterized by inferior displacement by gravity of intact nucleus within liquefied cortex. Histopathologically, cortical liquefaction is identified by the presence of Morgagnian globules, lens fiber fragments, and cortical clefts.16,109
Membranous cataracts represent varying stages of reabsorption of the lens. They can occur from trauma, congenital infection, in association with other ocular anomalies (persistent hyperplastic primary vitreous [PHPV], aniridia), or may be a part of broader syndromes.109
Lamellar or zonular cataracts are very common. They are characterized by focal opacification of lens fibers, sandwiched between the clear nucleus and cortex. Lamellar cataracts represent an insult to the lens at a specific point in the development of the affected group of lens fibers. They are usually inherited in an autosomal dominant fashion and have been mapped to chromosomes 1q and 13q.109,110
Nuclear cataracts present as the opacities in the embryonic or fetal nucleus (Fig. 2.10). Isolated nuclear cataracts are usually inherited as an autosomal dominant trait. They have also been associated with other ocular anomalies (e.g., microphthalmos) and infections (e.g., congenital rubella).109,110
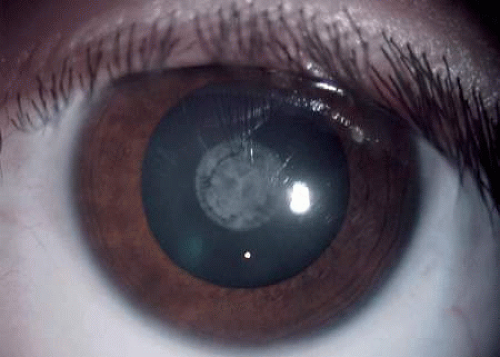
Figure 2.10. Several congenital cataracts are present in this eye. Sutural cataract appears as a Y-shaped opacity along the embryonic sutures. Central lenticular opacity (nuclear cataract) is also observed.
Cerulean or coronary cataracts appear as bluish opacities concentrated in the equatorial portion of the lens. They have autosomal dominant inheritance and have been mapped to chromosomes 17q and 22q.109,110
Anterior polar cataracts occur as central white opacities on the anterior surface of the lens. They may represent abnormalities of lens vesicle detachment or may be associated with abnormalities in pupillary membrane regression. Most are autosomal dominantly inherited. Histopathologically, anterior polar cataracts are similar to anterior subcapsular cataracts, displaying fibrous metaplasia of lens epithelial cells.16,109,110
Anterior pyramidal cataracts are relatively large, bilateral opacities that may extend anteriorly into the anterior chamber and, rarely, fuse with the cornea. They probably represent anomalies of lens vesicle detachment. Some cases may be secondary to intrauterine keratitis, whereby the lens becomes adherent to the inflamed cornea during fetal development. Familial anterior pyramidal cataracts display autosomal dominant inheritance.109,110
Histopathologically, anterior pyramidal cataracts may be indistinguishable from acquired anterior subcapsular cataracts, and are characterized by reduplication of the lens capsule, that surrounds a polar opacity composed of spindle-shaped lens epithelial cells and collagenous fibrous tissue (Fig. 2.11).16
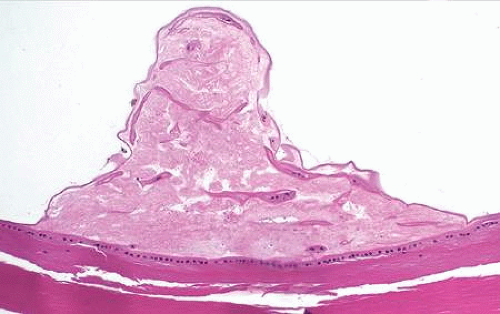
Figure 2.11. Pyramid-shaped anterior lenticular opacity protrudes into the anterior chamber. This pyramidal cataract is characterized by reduplication of the lens capsule that encloses abundant fibrous collagenous tissue and focal collections of lens epithelial cells (stain, hematoxylin-eosin [H&E]). (Courtesy of Harry Brown, MD.)
Sutural cataracts are very common. Sutural cataracts present as opacification along the embryonic sutures (Fig. 2.10). They are usually bilateral and familial, with autosomal dominant or X-linked recessive inheritance.109,110
Mittendorf’s dot is a small gray-white, slightly inferonasaly displaced, central posterior lenticular opacity. It represents the remnant of the anterior end of the hyaloid artery. Mittendorf’s dot is usually encountered as an isolated anomaly, but rarely may be associated with posterior lenticonus and persistent fetal vasculature.109
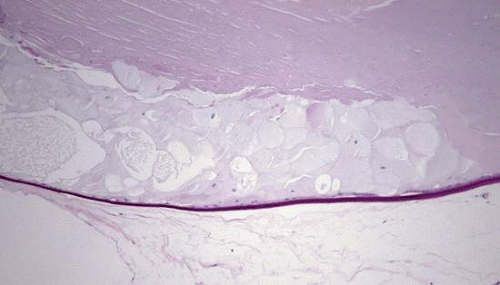 Figure 2.12. Posterior polar opacity is characterized histopathologically by posterior migration of enlarged equatorial lens cells (bladder cells of Wedl). (stain, periodic acid-Schiff [PAS])
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|