Congenital and Developmental Anomalies of the Orbit
Michael T. Yen
Patrick M. Flaharty
Richard L. Anderson
The various patterns of congenital facial and orbital anomalies have been recognized for many years. However, recent advances in gene mapping and surgical technique have dramatically changed our understanding as well as our approach to the management of these problems. Many congenital anomalies of the orbit are associated with more extensive defects that involve other structures of the face and skull. Although the orbital surgeon is well equipped to handle many localized problems of the orbit, more extensive anomalies require a multidisciplinary team, including craniofacial surgeons, neurosurgeons, pediatricians, orthodontists, ocularists, and others.
Congenital anomalies can affect the orbit in two ways. First, there can be a primary defect in the structural architecture of the bony orbit. This type includes defects of the anterior cranial base and facial skeleton. Alternatively, defects in the development of the globe and orbital soft tissues can induce secondary changes in the bony orbit. Moss and Salentijn’s theory of the functional matrix proposes an ongoing interdependence between the growth and development of orbital soft tissues and the surrounding bone.1
Most congenital and developmental anomalies of the orbit can be classified in one of three categories: localized anomalies of the orbit; craniosynostosis, or deformities of premature cranial suture closure; and facial clefting syndromes. Several other congenital anomalies, which do not fit into any of these categories, are discussed separately.
LOCALIZED ORBITAL ANOMALIES
Localized anomalies of the orbit and periorbital adnexa are the most common congenital defects seen in the routine practice of ophthalmology. These defects may affect other facial and intracranial structures, and it is extremely important for the ophthalmologist to identify the full extent of what may initially appear to be an isolated problem prior to any surgical intervention.
MICROPHTHALMOS AND ANOPHTHALMOS
True anophthalmos is a rare condition that results from failure of development or complete regression of the optic vesicle. This condition may be clinically indistinguishable from severe microphthalmos, which results from incomplete invagination of the optic vesicle or closure of the embryonic fissure. The term clinical anophthalmos has been used to describe patients who have no clinical or radiographic evidence of any ocular remnant, although true anophthalmos can only be verified after careful histologic sectioning of the orbital tissues.
Anophthalmos and microphthalmos are usually unilateral and may be associated with a variety of craniofacial and systemic anomalies, including orbital hypoplasia, facial clefts, basal encephalocele, hemifacial microsomia, mandibulofacial dysostosis, cardiac anomalies, polydactyly, and mental retardation. When they occur unilaterally, they also can be associated with anomalies of the contralateral “normal” eye, including cataract, cornea1 opacities, microphthalmos, coloboma, epibulbar dermoids, and nystagmus. Anophthalmos and severe microphthalmos frequently are associated with contracted conjunctival fornices, phimotic eyelids, and generalized hypoplasia of the periocular soft tissues (Fig. 1). When soft tissue contractures occur, the early use of conformers is essential to expand these tissues.2 This treatment should be instituted in the first month of life, with progressive enlargement of the conformer over time to achieve maximum expansion of the conjunctival fornix. Unfortunately, this treatment usually does not stimulate adequate orbital bone growth, and unilateral microphthalmos and anophthalmos may be associated with secondary orbital hypoplasia (Fig. 2). Serial implantation of progressively larger orbital implants or placement of expansile orbital implants has been advocated to stimulate bony orbital development.3,4
 Fig. 1. Microphthalmos of the left eye has resulted in secondary soft tissue contractures, including shortened fomices and small, phimotic eyelids. |
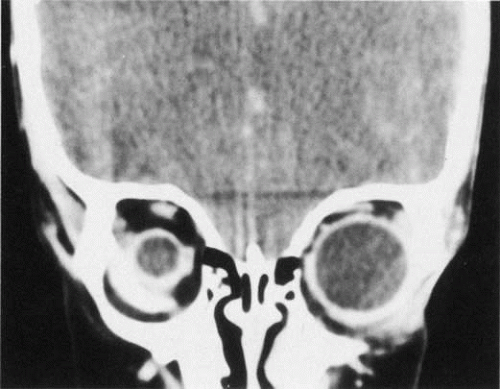 Fig. 2. Abnormal globe development for any reason may result in secondary orbital hypoplasia. Here, microphthalmos of the right globe is associated with relative orbital hypoplasia. |
CYSTIC ANOMALIES OF THE ORBIT
Many congenital cystic structures may arise from or involve the orbit. Some cystic structures, such as meningoencephaloceles or mucoceles, result from defects in the bony sutures of the cranial skeleton, allowing herniation of adjacent structures into the orbit. Other cystic structures, such as dermoid cysts, teratomas, and epithelial cysts, result from developmental anomalies of the orbital soft tissues. Most isolated orbital cysts have a subtle clinical presentation at birth, although some may present with extreme proptosis (Fig. 3). Ultrasonography can aid in the prenatal detection and monitoring of large orbital cysts.5
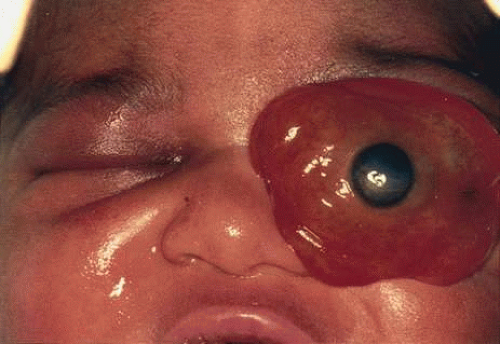 Fig. 3. Extreme proptosis at birth caused by a congenital orbital cyst. |
Dermoid cysts are the most common congenital orbital anomalies and represent developmental choristomas that are believed to arise from ectodermal nests pinched off by the fusion of bony sutures around the orbit. These cysts often originate from the frontozygomatic suture temporally but can also be seen nasally, arising from the frontonasal and frontolacrimal sutures; they rarely occur deep in the orbit.6 They commonly present during the first decade of life as a well-circumscribed, firm, rubbery subcutaneous mass just below the temporal eyebrow. Deeper dermoids can remain asymptomatic for many years, often presenting later in life as a slowly expanding orbital mass. Complete excision of these encapsulated lesions is the preferred treatment. Rupture of the cyst from trauma or during surgery may result in severe orbital inflammation (Fig. 4).
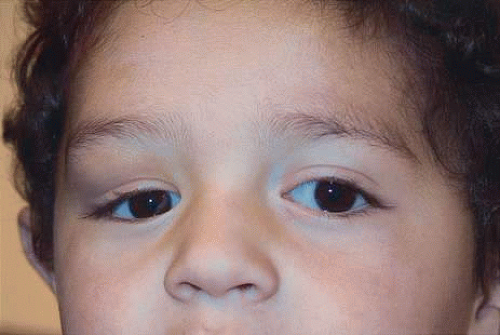 Fig. 4. A ruptured orbital dermoid cyst on the right side has caused swelling of the brow and upper eyelid as well as inferomedial displacement of the globe. |
CRANIOSYNOSTOSIS
Craniosynostosis implies premature fusion of the bony sutures of the skull. These craniofacial anomalies usually display a sporadic inheritance pattern, although several well-recognized syndromes have distinct inheritance patterns, such as the autosomal dominant pattern of Crouzon’s disease. Although premature suture fusion was believed to be the primary pathologic process, genetic mapping and molecular studies suggest that this early fusion may be a result of altered cytokine and extracellular matrix component expression.7
The phenotypic pattern of deformity is a direct result of the sutures involved (Fig. 5). Scaphocephaly is an elongated, narrow cranium associated with premature fusion of the sagittal suture. Brachycephaly is a short, wide cranial vault associated with bilateral coronal synostosis. Plagiocephaly results from premature closure of one coronal suture, leading to prominent orbital asymmetry (Fig. 6). A flattened, recessed forehead occurs on the affected side, and persistent growth of the contralateral side results in frontal bossing, inferolateral orbital dystopia, and a prominent occiput. This deformity is reminiscent of hemifacial microsomia. Trigonocephaly is a triangular deformity of the anterior cranial fossa that results in medial displacement of the orbits (hypotelorism) (Fig. 7). Acrocephaly results from multiple suture closure, including bicoronal synostosis. Typically, there is excessive skull height and a pointed head.
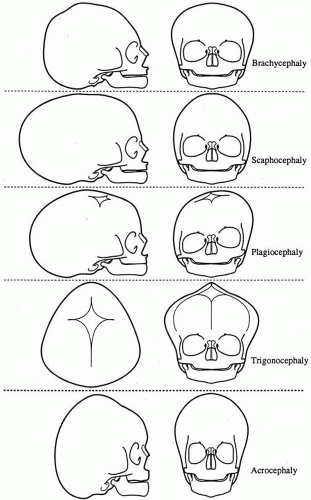 Fig. 5. The skull deformity in cranlosynostosis is a direct result of the sutures involved. |
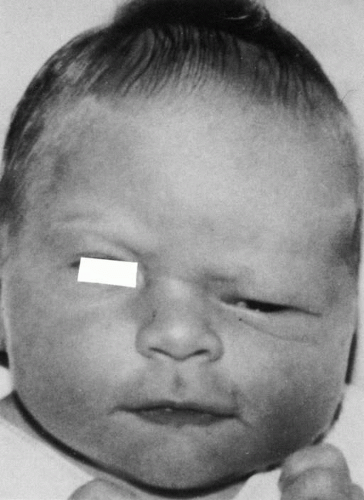 Fig. 6. Unilateral coronal synostosis results in severe orbital asymmetry (ptogiocephaly). The uninvolved side (left) shows dramatic growth relative to the prematurely fused side (right)). |
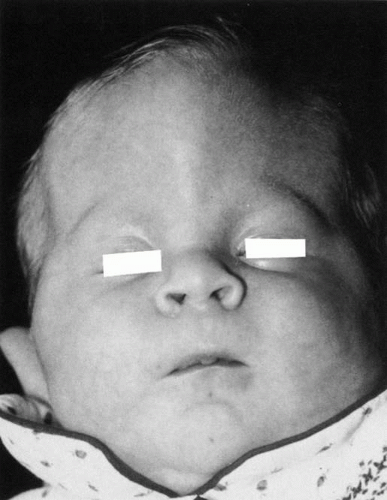 Fig. 7. Trigonocephaly or triangular deformity of the anterior cranial fossa, with associated hypotelorism. |
CROUZON’S DISEASE
In 1912, Crouzon described the froglike facies characteristic of this distinct anomaly, and his name has been associated with this syndrome ever since (Fig. 8). Most cases of this autosomal dominant disease have been mapped to the fibroblast growth factor receptor 2 gene located on chromosome 10.8 Ocular findings include exophthalmos, hypertelorism, strabismus, extraocular muscle agenesis or anomaly, nystagmus, papilledema, and optic atrophy. A variable pattern of suture closure is seen, including coronal, sagittal, and lambdoid sutures. The deformities of the orbit and cranial vault are in part a result of the compensatory expansion of the cranium from increased intracranial pressure. Forward displacement of the greater wing of the sphenoid bone results in a shortening of the lateral orbital wall and a dramatic reduction in orbital volume. To compound the problem, there is also inferior displacement of the orbital roof from anterior cranial fossa expansion and shortening of the orbital floor from maxillary hypoplasia. Tessier estimated that these defects account for a 6-cc reduction in orbital volume, or approximately 20% to 25% of the total volume of the orbit.9
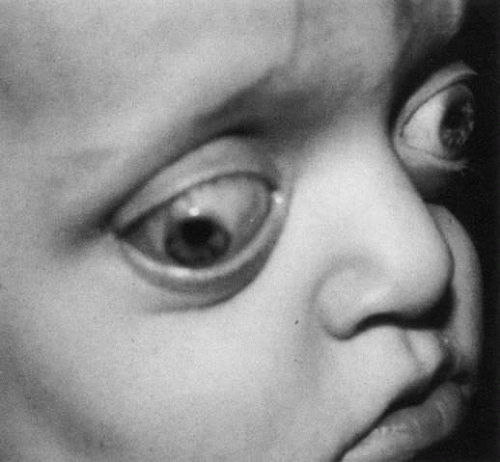 Fig. 8. The froglike facies of Crouzon’s disease results from maxillary hypoplasia, shallow orbits, and prominent exophthalmos.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|