2 Congenital and Developmental Anomalies
INTRODUCTION
Congenital anomalies are developmental disorders that are present at birth. Congenital malformations are caused by chromosomal abnormalities, mutant genes, and major environmental factors such as infections, drugs and toxins, or radiation. In many instances, the cause is unknown. Heritable disorders are genetically determined and may or may not affect the phenotype. Heritable traits may be manifest at birth, or they may become obvious later in life.
Etiologic factors that act early during embryogenesis (conception to 2 weeks) or the initial stages of ocular organogenesis (2 weeks to 3 months) tend to have profound effects on ocular development. The development of the eye commences about 24 days after fertilization when the optic pits form in the anterior part of the embryo’s neuroectodermal plate. The pits subsequently evaginate to form the optic vesicles, which invaginate to form the optic cups 4 days later. The neuroectoderm comprising the inner layer of the optic cup is destined to form the neurosensory retina, the nonpigmented ciliary epithelium, and the posterior layer of iris epithelium, while the outer layer of the optic cup gives rise to the retinal pigment epithelium (RPE), the pigmented ciliary epithelium, and the anterior layer of iris pigment epithelium, which includes the dilator muscle. The optic vesicles induce the formation of the lens placodes in the overlying surface ectoderm. The lens placodes invaginate to form the lens vesicles as the optic cups form. Transitory fissures develop in the outer wall of the optic cups on day 29. These embryonic fissures give the hyaloid arteries access to the interior of the developing eye. The fissures subsequently are obliterated by fusion, which usually is complete by the 6th week of gestation. Abnormalities affecting each of these events can become manifest as severe developmental anomalies.
Primary anophthalmos is a rare anomaly caused by failure of optic vesicle evagination. Primary anophthalmos usually is bilateral and occurs sporadically in otherwise healthy individuals. Secondary anophthalmos is caused by suppression of the entire anterior part of the neural tube, which is lethal. In consecutive anophthalmos, the optic vesicle evaginates and then undergoes degeneration. Histologic examination of the orbit usually discloses rudimentary neuroectodermal structures, which are absent in primary anophthalmos.
SYNOPHTHALMIA/CYCLOPIA
Synophthalmia/cyclopia is a striking anomaly caused by a failure of formation or induction of the anterior neural tube including the eye fields (Fig. 2-1A,B). Although it is often called a fusion anomaly, synophthalmia/cyclopia actually reflects a failure of complete bicentricity to emerge. The disorder is a continuum of anomalies that involves the brain, nose, orbits, and bones in addition to the eyes. Bicentricity fails to emerge at an early stage; cells that are induced as eye tissue complete all stages of development with a high degree of fidelity. The greatest degree of ocular differentiation occurs anteriorly and laterally; there is duplication of anterior ocular structures and fusion posteriorly with a single optic nerve. A rudimentary nose or nasal proboscis is present above the single midline orbit (Fig. 2-1A). The proboscis is caused by faulty migration of the frontonasal processes. Failure of bicentricity also involves the forebrain, invariably producing a malformation called holoprosencephaly, which is marked by failure of the brain to divide into right and left hemispheres. Most cases of this clinical spectrum have synophthalmia; true cyclopia is exceedingly rare. The lethal malformation usually is sporadic, but it may be a manifestation of trisomy 13. Dominantly and recessively inherited familial cases have been reported. Holoprosencephaly has been linked to mutations in a number of genes including the human sonic hedgehog gene on chromosome 7q36 and the SIX3 sine oculo hemeobox gene on chromosome 2p21. Pregnant ewes who ingest toxic alkaloids from the false hellebore plant Veratrum californicum on the 14th or 15th day of gestation give birth to cyclopic lambs.
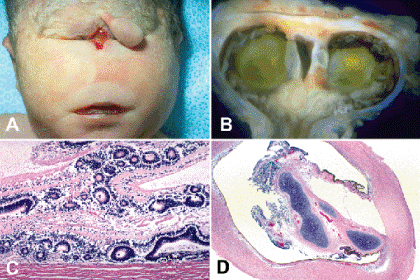
Fig. 2-1. Trisomy 13. A. Synophthalmia, trisomy 13. A pedunculated nasal proboscis is located above the single midline orbit. B. Synophthalmia, trisomy 13. Small, synophthalmic eye from infant shown in Figure 2-1A has two lenses and a single optic nerve. Septum between lenses contains two foci of hyaline cartilage. Mass of dysplastic retina fills vitreous cavity. C. Coloboma and retinal dysplasia, trisomy 13. Mass of disorderly dysplastic retina containing large multilayered rosettes adheres directly to the sclera within the coloboma. The choroid is absent. D. Intraocular cartilage, trisomy 13. Mesenchymal tissue filling coloboma surrounds a focus of hyaline cartilage in severely microphthalmic eye.(C. Hematoxylin-eosin (H&E), ×25, D. H&E, ×10)
Congenital cystic eye is a very rare anomaly caused by complete failure of optic vesicle invagination. Partial arrest in vesicle invagination probably causes extreme microphthalmos, which may simulate anophthalmos clinically. Congenital nonattachment of the retina is caused by faulty invagination of the optic cup and failure of its inner and outer layers to meet posteriorly.
Uveal colobomas are developmental anomalies caused by faulty closure of the embryonic fissure. Derived from the Greek word for “mutilation,” coloboma is defined as “a condition where a portion of the structure of the eye is lacking.” Colobomas typically are located inferonasally in the territory of the fissure. They can involve the iris, ciliary body, choroid, or any combination of the three, and also may affect the optic nerve. Because the primary defect in a typical coloboma is in the neuroectoderm, absence of the mesectodermal uveal stroma is a secondary phenomenon. Occasionally, the uveal tissue undergoes dysplasia or metaplasia forming cartilage, muscle or fat. An absolute scotoma is present in the region of the coloboma because the overlying retina is absent or dysplastic.
Although most colobomas are sporadic, they occasionally are inherited as isolated ocular defects, usually in an autosomal dominant fashion with incomplete penetrance. Colobomas occur in infants with CHARGE syndrome and the Cat Eye, Kabuki, Wolf-Hirschhorn (4p–), and 13q deletions syndromes as well. In addition to colobomas, infants with CHARGE syndrome have congenital heart defects, choanal atresia, mental retardation, and genital and ear anomalies. CHARGE syndrome is associated with mutations or complete deletion of the CHD7 (helicase DNA-binding protein-7) gene on chromosome 8q12. Colobomas also are characteristic findings in several syndromes caused by the duplication or deletion of chromosomes or chromosomal fragments including trisomy 13. Colobomas in severely microphthalmic (<10 mm) eyes from infants with trisomy 13 often contain foci of hyaline cartilage and dysplastic retina (Fig. 2-1C,D).
Coloboma with cyst is a severe form of embryonic fissure anomaly characterized by a cystic outpouching of ectatic sclera that communicates with the interior of the eye through a posterior coloboma (Fig. 2-2). The intraocular contents protrude outward through the fissure, and the cyst is lined by a layer of atrophic or dysplastic neuroectodermal tissue. The cyst may become much larger than the eye (microphthalmos with cyst).
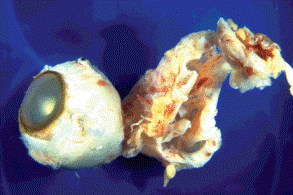
Fig. 2-2. Microphthalmos with cyst. Cystic outpouching of ectatic sclera is larger than microphthalmic eye. The lumen of the cyst was lined by atrophic neuroectodermal tissue and it communicated with the interior of the eye through an optic nerve coloboma. The cyst ruptured intraoperatively.
Atypical colobomas are not related to closure of the embryonic fissure and can occur anywhere. Macular colobomas probably result from intrauterine infections such as congenital toxoplasmosis. Colobomas of the eyelid occur in Goldenhar syndrome.
Optic nerve aplasia usually is unilateral and occurs sporadically in individuals who have no systemic abnormalities. The optic nerve is absent and the retina is avascular and lacks ganglion cells and axons.
Malformations that are localized to a single ocular structure usually are related to damage that occurs during the fetal period of ocular development (3rd to 9th month). Most are discussed in the chapters on individual ocular structures that follow.
Ocular malformations occur in a number of chromosomal duplication or deletion syndromes. Down syndrome (trisomy 21) is the most common chromosomal syndrome and the most common cause of mental retardation. The “mongoloid” appearance of patients with Down syndrome is caused by an upward and outward slanting of their palpebral fissures, which are almond shaped. Other ocular findings include epicanthal folds, significant refractive errors, especially high myopia, cataract, and strabismus, usually esotropia, which occurs in approximately 40%. Congenital ectropion, iris hypoplasia, keratoconus with acute hydrops, and an increased number of retinal vessels crossing the optic disc margin also occur.
Brushfield spots appear as a concentric ring of white or yellowish spots on the anterior surface of the iris in 85% of blue- or hazel-eyed patients with Down syndrome (Fig. 2-3). Brushfield spots are focal condensations of stromal collagen that are visible through the transparent anterior border layer of the blue iris, and tend to be accentuated by concurrent iris atrophy. Discrete stromal condensations that resemble Brushfield spots called Kruckmann-Wolfflin bodies occur in some normal persons with blue or hazel eyes.
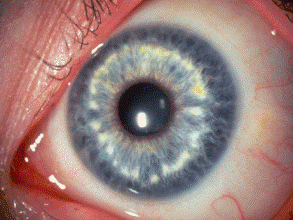
Fig. 2-3. Brushfield spots, trisomy 21. Focal stromal condensations form a ring of white spots in the middle third of the iris in a blue-eyed patient with Down Syndrome. (Photo courtesy of Dr. Edward A. Jaeger, from Eagle RC Jr. Congenital, developmental and degenerative disorders of the iris and ciliary body. In: Albert DM, Jakobiec FA, eds. Principles and Practice of Ophthalmology. Clinical Practice, vol. 1. Philadelphia, PA: Saunders, 1993:367–389.)
Severe ocular malformations including anophthalmos, synophthalmia/cyclopia, microphthalmia, PHPV, retinal dysplasia, colobomas, and intraocular cartilage occur in infants with trisomy 13 (Patau syndrome) (Fig. 2-1A–D). Affected infants have cleft lips and palates, cardiac and pulmonary defects, arrhinencephaly and holoprosencephaly. Few survive the first year of life.
Epicanthal folds, ptosis, blepharophimosis, microphthalmos, corneal opacities, and congenital glaucoma are found in trisomy 18 (Edwards syndrome), the second most common autosomal trisomy.
Retinoblastoma occurs in the 13q– syndrome if the deletion includes the q1-4 band. The 22q+ syndrome includes microphthalmia, the posterior ulcer of von Hippel, and severe retinal dysplasia. Ocular abnormalities have been reported in the 18p–, 18q–, 18 ring chromosome, 5p– (cri-du-chat), and the 4p– (Wolf-Hirschhorn) syndromes.
HERITABLE DISORDERS WITH OCULAR MANIFESTATIONS
A variety of heritable disorders caused by genetic mutations have ocular manifestations.
Most cases of aniridia are caused by mutations in the PAX6 gene on the short arm of chromosome 11. The term aniridia is a misnomer; most cases have severely hypoplastic irides that are hidden clinically by opaque limbal tissues (Fig. 2-4). Aniridia is a spectrum of ocular disease that also includes foveal and optic nerve hypoplasia, cataract, secondary glaucoma, and corneal opacification. Eighty-five percent of aniridic patients have autosomal dominant familial aniridia, which is an isolated ocular defect. The association of sporadic aniridia and Wilms tumor or nephroblastoma is called Miller syndrome and accounts for about 13% of cases. Miller syndrome is caused by deletions in the short arm of chromosome 11 that include both the PAX6 gene and another closely linked tumor suppressor gene (WT1) involved in the pathogenesis of Wilms tumor. The PAX6 gene plays a central role in ocular development throughout the animal kingdom. PAX6 mutations have been identified in patients with other anterior segment malformations including Peters anomaly and autosomal dominant keratitis.
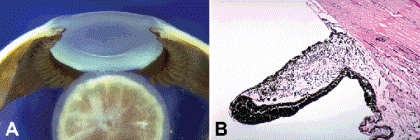
Fig. 2-4. Aniridia. A. Overhang of opaque limbal tissue hides skirt of hypoplastic iris in peripheral anterior chamber. The lens dislocated during sectioning of eye obtained postmortem from adult with familial aniridia. B. Stubby hypoplastic iris leaflet from eye seen grossly in (A) has thickened pigment epithelium and lacks sphincter and dilator muscles. (H&E, ×25)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


