Comprehensive Cleft Care
Alexander J. Davit III
Todd Otteson
Joseph E. Losee
Despite the numerous advances in caring for patients affected with cleft lip and/or palate (CL/P), parents who learn their child is affected can often feel considerable anxiety and confusion over the significance of this diagnosis. In order to better care for cleft patients and their families, treatment protocols have evolved that utilize a multidisciplinary team-based approach, incorporating skills offered by several subspecialties, including otolaryngology, plastic surgery, orthodontics, pediatric dentistry, speech pathology, audiology, psychology, and social services. Each discipline is focused on delivering coordinated, comprehensive care to the cleft patient.
Numerous authors have written extensively on this challenging subject; this chapter is intended to serve as a detailed outline for the evaluation and treatment of patients affected with CL/P, focusing on the salient points of comprehensive cleft care. At the Children’s Hospital of Pittsburgh of UPMC, our cleft team focuses on every stage in each patient’s development where intervention may be considered to benefit the patient. It is important to recognize that caring for the child with a cleft is a long-term process, extending from infancy into early adulthood, and is a journey with several important “stops” along the way. During the child’s first year of life, the primary focus of care is in promoting proper nutrition and growth, while also performing the initial soft tissue reconstructions. Repairing the CL/P in a skilled manner is of utmost importance, thereby laying the foundation for the patient’s future facial appearance, but it is hardly the final procedure for most patients today. After infancy, a major focus is on speech outcomes, when selected patients may benefit from revisional speech surgery to address velopharyngeal insufficiency. During the period of mixed dentition, the patient is evaluated for alveolar bone grafting and “touch up” revisional soft tissue procedures to the lip and nose. In late adolescence, patients are considered for orthognathic surgery and definitive rhinoplasty to refine the patient’s “final result.” During this entire process, the cleft team routinely follows patients in order to assess their individual needs with regard to appearance, psychosocial development, speech, hearing, orthodonture, and dentistry. Comprehensive team care of the cleft patient can be a clinically gratifying endeavor, one that necessitates a multidisciplinary approach to deliver reproducible, high-quality results for the patient.
EPIDEMIOLOGY: INCIDENCE AND GENETIC FACTORS
CL/P is one of the most common congenital anomalies: the prevalence in North America is currently 0.2 to 2.3 cases per 1,000 population, while the prevalence of isolated cleft palate (CPO) is 0.1 to 1.1 cases per 1,000 (1, 2). For CL/P, these rates vary between ethnic subgroups: 3.6 per 1,000 in Native Americans, 2.1 per 1,000 in Asians, 1 in 1,000 in Caucasians, and 0.41 per 1,000 in African Americans (1, 3, 4). CPO has a relatively stable incidence across ethnic subgroups (1). The male to female ratio for CL/P is 1.5 to 2.0:1 (2). For CPO, the converse is true; females are more commonly affected, with a male to female ratio of 1:2 (2).
Development of CL/P or CPO is believed to be a complex, multifactorial process involving multiple risk factors, genetic mutations, family history of orofacial clefting (OFC), maternal diseases and behaviors, fetal exposure to teratogenic medications, and nutritional deficiencies or excesses. A positive family history of OFC has long been recognized as a strong risk factor and was first studied in 1942, when Fogh-Anderson began to apply statistical analysis to familial inheritance patterns of CL/P (1). Genetic studies in both humans and mice have identified transforming growth factor beta 3 (TGFβ3) and msh homeobox 1 (MSX1) as genes that play an important part in the development of CL/P (1).
While most cases of CL/P and CPO are nonsyndromic, there are a multitude of genetic syndromes that may be associated with CL/P and CPO. The most common syndrome associated with CL/P is Van der Woude syndrome, an autosomal dominant syndrome characterized by blind lower lip pits in addition to CL/P (5). The most common syndromic diagnoses associated with CPO are the autosomal dominant syndromes caused by microdeletions or additions of chromosome 22q11.2: DiGeorge syndrome, conotruncal anomaly face syndrome, and velocardiofacial syndrome; these have an incidence rate of 1:4,000 live births (5, 6). Other syndromes associated with CPO include Stickler syndrome, popliteal pterygium syndrome, and ectrodactyly ectodermal cleft syndrome (5).
Environmental factors are also believed to play a pivotal role in the development of CL/P. These factors can be grouped into several categories: maternal health, teratogenic substances, and nutritional factors. The association between birth defects and maternal illnesses such as diabetes mellitus and gestational diabetes has been studied extensively; an increased incidence of congenital malformations is notably higher in patients with diabetes mellitus, but not gestational diabetes (2). Maternal obesity has also been studied as a risk factor for CL/P and CPO, and women with a BMI greater than 29 kg/m2 have been shown to have a 1.3 times higher risk of having a child with a cleft when compared to women with nonobese BMI values (2). Teratogenic medications associated with development of CL/P include valproic acid, phenytoin, retinoic acid, dioxin, and thalidomide (1). Maternal tobacco smoking has been implicated in the development of OFC: CL/P and CPO are approximately 1.2 times more likely than in the general population (2). Although the children of women who consume heavy amounts of alcohol during pregnancy are known to be at increased risk for congenital malformations, the specific relationship between the development of OFC and maternal alcohol consumption remains unknown (2). Based on previous animal studies, maternal folic acid supplementation has frequently been considered to have a protective effect with regard to CL/P, although in a case-control study in the United Kingdom, higher maternal folic acid intakes were not associated with a lower incidence of OFC (7).
EMBRYOLOGY
Embryologic formation of the nose and upper lip begins during the fourth week of gestation, when the bilaminar prechordal plate containing ectodermal and endodermal elements is formed. This structure gives rise to the stomodeum, or primitive mouth, about which five facial prominences develop: the frontonasal prominence, paired maxillary prominences, and paired mandibular prominences (8).
Development of the upper lip and nose is controlled by several developmental pathways, including sonic hedgehog (SHH), wingless type (Wnt), bone morphogenic protein (BMP), and fibroblast growth factor (FGF) (9). Fusion of the frontonasal and maxillary processes begins to evolve in the anterior face at approximately 32 days’ gestation, when the ventrolateral aspects of the frontonasal prominence undergo cellular proliferation to form the nasal placodes, which in turn give rise to the medial and lateral nasal processes (9). During this process, the maxillary processes also undergo rapid mesenchymal growth, and at the 38th day of gestation, fusion commences between these processes and the lateral and medial nasal prominences, approximating the distal tips of the medial and lateral nasal processes, eventually causing contact and fusion between them (9, 10) (Fig. 103.1). Further development of the upper lip and nose follows, with the continued growth of the maxillary processes causing anteromedial positioning of the nasal pits, which undergo further maturation into the nasal ducts (9). Upper lip development is typically complete by the 48th day of gestation, once the interposed epithelial elements have undergone apoptosis and the remaining mesenchymal elements undergo final fusion between the maxillary and medial nasal prominences, to be followed by anteroposterior formation of the primary and secondary palate (9).
Palatal anatomy is subdivided into the primary and secondary palates, with the primary palate being formed by the frontonasal prominence and the secondary palate being formed by the paired lateral maxillary prominences (8). In the eighth week of normal embryologic development, the tongue begins to be withdrawn from its position between the lateral maxillary prominences, and they begin to shift from a vertical to horizontal orientation, ultimately undergoing fusion in the normal fetus (8). Several different mechanisms are believed to contribute to this vertical migration, including increased mesenchymal proliferation, increased tissue fluid content, and multiple signaling pathways, including platelet-derived growth factor, FGF10, SHH, and TGFβ3 (8). Once the palatal shelves meet in the midline, the line of fusion forms in the midpoint of the hard palate, later progressing both anteriorly and posteriorly from this point (8).
PRENATAL DIAGNOSIS AND COUNSELING
Prenatal diagnosis of CL/P, along with other developmental anomalies, has become more common with the widespread use of screening ultrasonography. It is important to recognize that approximately 50% of children with cleft lip and alveolus have other anomalies as well; 22% have an abnormal karyotype (11). In several large studies, it was possible to identify an isolated cleft lip deformity by ultrasound approximately 30% to 55% of the time at 24 weeks’ gestation (11, 12). The high variability in detection rates is believed to be due to differences in each center’s ultrasonographic examination protocols (12). The ability to identify CPO on ultrasound is significantly lower,
with a rate of 1.4% in the Eurofetus study (11). Detection rates with newer imaging modalities, such as 3-D ultrasound and MRI, have yet to be fully elucidated at this time.
with a rate of 1.4% in the Eurofetus study (11). Detection rates with newer imaging modalities, such as 3-D ultrasound and MRI, have yet to be fully elucidated at this time.
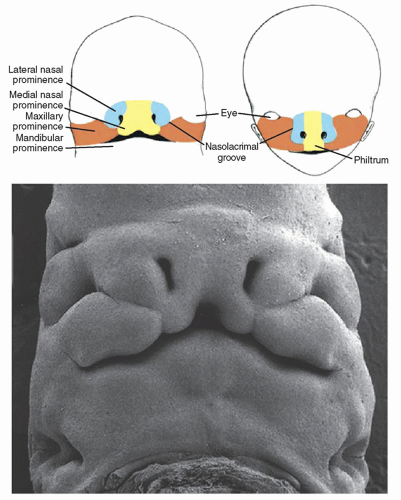 Figure 103.1 Facial development in the embryo. Note the development of the upper lip, philtrum, and nose from the maxillary, medial nasal, and lateral nasal prominences. (From van Aalst J, Kolappa KK, Sadove M. Nonsyndromic cleft palate. Plast Reconstr Surg 2008;121(1):1-14, with permission.) |
The goal of detecting OFC in the prenatal period is to allow parents to obtain appropriate genetic counseling, emotional support, and treatment planning. In counseling parents with regard to CL/P, it is important to discuss the potential recurrence risk, so informed choices with future family planning may be made. Population studies have determined the statistical risk of having a second child with CL/P or CPO. In first-degree relatives of patients with a nonsyndromic OFC, the risk of having a child with CL/P is approximately 3% to 4% (13). For patients affected with CPO, the risk for first-degree relatives is approximately 1.8% (13). For an affected parent with CL/P who already has one affected child, the recurrence risk is approximately 10% to 17% (5, 8). For an affected parent with CPO who already has one affected child, the risk is slightly lower, 8% to 15% (13) (Table 103.1).
TABLE 103.1 FAMILIAL RISKS FOR NONSYNDROMIC CLEFT LIP AND PALATE | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||
FEEDING THE CLEFT PATIENT
Establishing a reliable oral feeding strategy is critical for patients born with OFC. Patients with wide, complete clefts often have significant initial challenges with oral feeding, since the cleft prevents them from making an adequate seal on the bottle or breast (14). This can lead to inefficient suckling, resulting in the infant tiring more readily. Because of the ineffectual seal, patients often swallow a significant amount of air during feeds and require more aggressive burping during and after feeding. Most cleft patients will be able to feed well orally with the assistance of bottles with softer nipples and/or a valve flow control system. In addition, modified positioning that places the infant’s head in a more vertical position helps to avoid significant reflux and air swallowing in many patients (14). Specialized nipple and bottle kits used for feeding cleft patients include the Mead-Johnson Cleft Palate Feeder, the Pigeon Cleft Palate Nipple and Bottle, and the Special Needs Feeder (Haberman Feeder). These special bottles can be made available and demonstrated to the cleft patient’s parents well in advance of their due date—during their prenatal educational appointment (14). Prenatal counseling regarding the use of these techniques is important for families to be well prepared for the challenges of feeding the cleft patient. Cleft patients without other medical issues requiring hospitalization are discharged home once able to ingest appropriate quantities of breast milk or formula to prevent malnutrition and dehydration, and the family is instructed to follow up in the cleft clinic in the week after discharge. Patients should return to their birth weight by their 2-week postnatal examination and should thereafter gain approximately 0.5 to 1 oz./day (14). Frequent reevaluations early in the patient’s life ensure that the patient is monitored for any signs or symptoms of malnutrition or dehydration, which may require medical intervention.
ANATOMY AND CLASSIFICATION OF THE CLEFT DEFORMITY
Unilateral Cleft Lip Anatomy
The anatomy of the upper lip and nose is complex and involves several important structures: the nasal alae, the columella, the nasal septum, the philtrum of the upper lip, the wet and dry vermillion, the orbicularis oris muscles, and the oral commissures. These structures are affected differently in patients with unilateral and bilateral clefts. In the unilateral cleft, there is an imbalance of the lip/nasal morphology, causing the medial premaxillary segment to rotate externally and upward, while also causing internal and posterior rotation of the lateral minor maxillary segment (5). The orbicularis oris muscle is abnormally inserted into the anterior nasal spine at the base of the columella on the nonclefted side and the nasal alar base on the clefted side (5). The clefted side’s lower lateral cartilage is typically flattened and displaced posterolaterally, and the middle crus (dome) of the lower lateral cartilage is separated from the noncleft middle crus (5). The caudal septum is frequently dislocated from the vomerine groove and displaced into the nonclefted nostril (5).
Bilateral Cleft Lip Anatomy
In patients with a complete bilateral cleft lip, the premaxillary segment is positioned more anteriorly than in nonclefted patients, due to its lack of fusion with the lateral maxillary segments. The prolabium does not contain orbicularis oris muscle, and the vermilion and white roll are significantly deficient. The nasal deformity is fairly uniform and consists of flared nasal alar bases, a wide and flat nasal tip, and a short columella (5). Incomplete bilateral clefts have similar features, although the protrusion of the premaxillary segment is less severe than that of patients with a complete deformity.
Palatal Anatomy
The palate may be divided into the primary palate and the secondary palate; the primary palate (premaxilla) is located anterior to the incisive foramen, and the secondary palate located posterior to the incisive foramen (15). The secondary palate is further subdivided into the hard palate posterior to the incisive foramen and the velum, or soft palate. The velum contains five paired muscles: the muscularis uvulae, the palatoglossus, the palatopharyngeus, the levator veli palatini (LVP), and the tensor veli palatini (TVP) (16). The muscularis uvulae originates anteriorly on the tensor aponeurosis in the midline and extends posteriorly in the midline to the uvula (16). The palatoglossus muscle originates from the dorsolateral aspect of the tongue and extends within the anterior tonsillar pillar to insert into the velum (15, 16). The palatopharyngeus muscle originates from the superior pharyngeal constrictor and extends within the posterior tonsillar pillar to insert into the velum, dividing into a superior oral head and an inferior nasal head, both enveloping the LVP muscle within the velum (16). The TVP originates from the eustachian tube (ET) and greater wing of the sphenoid, coursing anteriorly around the hook of the hamulus and inserting medially upon the tensor aponeurosis (15). In Huang’s anatomic dissection of the paratubal muscles, the LVP was found to originate from the junction of the bony and cartilaginous portions of the eustachian tube, rather than the quadrate region of the petrous temporal bone as previously described (17).
Clefts of the soft palate alter the ability of the velum to obturate the nasopharyngeal port and have a relatively predictable anatomic structure. Rather than coursing medially to coalesce in the midline of the middle third of the velum,
the paired LVP muscles follow a more anterior direction to insert upon the posterior edge of the hard palate. In patients with cleft palate, the LVP has three abnormal attachments: the superior pharyngeal constrictor, the tensor aponeurosis, and the posterior edge of the hard palate (15) (Fig. 103.2).
the paired LVP muscles follow a more anterior direction to insert upon the posterior edge of the hard palate. In patients with cleft palate, the LVP has three abnormal attachments: the superior pharyngeal constrictor, the tensor aponeurosis, and the posterior edge of the hard palate (15) (Fig. 103.2).
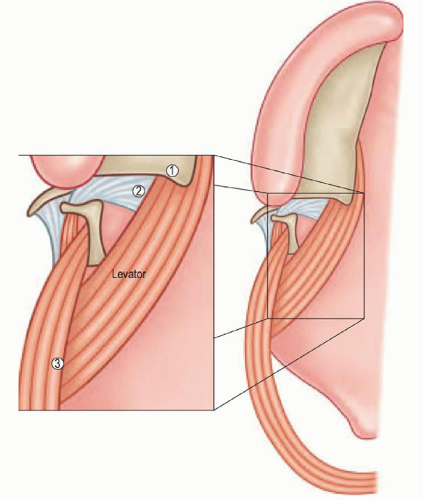 Figure 103.2 The three abnormal insertion points of the levator palatini muscle—1. posterior edge of the hard palate, 2. TVP aponeurosis, 3. superior pharyngeal constrictor muscle. (From Losee JE, Smith D. Cleft palate repair. In: Butler CE, ed. Head and neck reconstruction. Philadelphia, PA: Saunders Elsevier, 2009:275, with permission.) |
Anatomical Classification Systems
Multiple classification systems for CL/P have been proposed; each has its own merits and limitations. These systems range from simple lettering systems, such as Kriens’ LAHSHAL system, to Millard’s modification of Kernahan “striped-Y” (18). At our institution, we prefer to use the LAHSHAL system for clefts of the lip and alveolus, given its simplicity and ability to address complete, incomplete, and submucous cleft anatomy. The LAHSHAL descriptive system represents the patients Lip, Alveolus, Hard palate, Soft palate, Hard palate, Alveolus, Lip anatomically from right to left. It is convenient in that it allows the clinician to categorize the cleft with the use of capital letters to signify a complete cleft, small letters to signify an incomplete cleft, an asterisk to signify a submucous or microform cleft, and an “X” to signify a normal structure. Hence, a left-sided complete cleft of the lip, alveolus, hard palate, and soft palate would be: “XXXSHAL.” An incomplete right-sided lip and cleft of the soft palate would be: “lXXsXXX” (Fig. 103.3). For similar reasons, we utilize the Veau classification system for clefts affecting only the palate. The Veau classification system for cleft palate deformities is also simple and convenient: Veau I clefts involve only the soft palate; Veau II clefts involve the hard and soft palate; Veau III clefts are unilateral complete clefts of the alveolus, hard, and soft palate; and Veau IV clefts are bilateral complete clefts of the alveolus, hard, and soft palate. Often the Veau III and IV clefts are associated with clefts of the lip (15).
SURGICAL MANAGEMENT OF THE PRIMARY DEFORMITY: SEQUENCING AND TIMING
There is no “cookbook” approach to cleft care, and each patient requires an individualized treatment plan. As an example, patients presenting to large cleft centers may live several hours away. This distance may present a logistical barrier to the use of innovative presurgical treatments, like presurgical infant orthopedics (PSIO), that often require weekly visits for up to several months. If this is the reality, then prior to definitive surgery for the lip and nose, a lip adhesion procedure may be considered as an alternative to PSIO. While there is a general “timeline” for the overall surgical care of these patients, an individualized approach is preferred, taking into consideration the patient’s and family’s particular needs.
Presurgical Infant Orthopedics
Presurgical infant orthopedics (PSIO) is a treatment started early in a cleft patient’s life. The goal of PSIO is to realign the clefted maxillary shelves, bring the widely separated lip margins together, and reshape the nose, “setting-up” and facilitating the surgical reconstruction. PSIO modalities include active techniques such as the Latham appliances or passive techniques such as lip taping or nasoalveolar molding (NAM). NAM, a passive form of PSIO, has gained popularity among cleft centers and is generally started in patients with complete unilateral or bilateral clefts at approximately 1 week of age. Weekly evaluations with modification of the NAM device to mold the lip, alveolus, and nasal ala are continued until the patient is 3 to 4 months of age, immediately prior to definitive repair of the cleft lip/nose deformity. In patients with complete clefts who are not candidates for NAM, lip adhesion surgery may be performed at approximately 1 to 2 months of age. Originally described in 1960 by Johansson and Olson (1960), staged lip repair with lip adhesion as the initial procedure has been utilized in patients with wide complete clefts in order to convert




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
