Microbial Inflammation
Microbial infections of the orbit develop due to a variety of organisms, which are introduced to the orbit by different routes. Bacterial orbital infections are the most common, which primarily originate from the paranasal sinuses, and to a lesser degree secondary to penetrating injuries with or without foreign bodies, and rarely, secondary to endophthalmitis or systemic infectious process such as bacterial septicemia and tuberculosis. The most common cause of bacterial orbital cellulitis is
Staphylococci and
Streptococci species. Although
Haemophilus influenza was a common causative organism in children younger than 4 years, recent vaccination efforts have decreased the incidence of
H. influenza cellulitis significantly.
57 On the other hand, community-acquired methicillin-resistant
Staphylococcus aureus (MRSA) has been increasingly reported as a vision-threatening pathogen to afflict immunocompetent children and adults.
58Anatomically speaking, if the infection is limited to anterior of the orbital septum, involving the eyelids, it manifests itself primarily with lid and conjunctival edema and some venous congestion but not with proptosis.
59 This process, which is known as
preseptal cellulitis, is usually initiated with skin injuries or an upper respiratory tract infections in children. On the other hand, if the infectious process involves orbital soft tissues beyond the septum, it is called
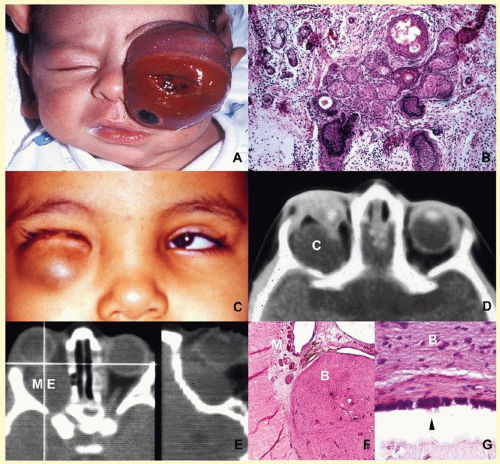
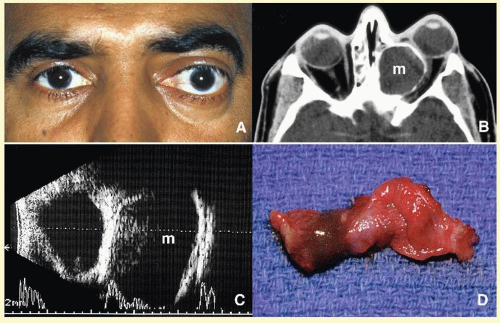
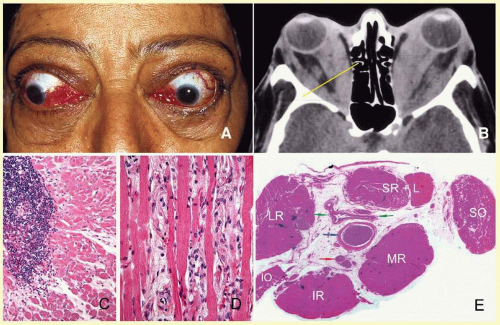
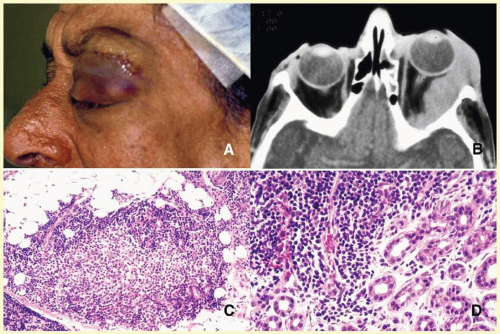
orbital cellulitis (
Fig. 17.6). Cellulitis usually develops secondary to a paranasal sinus disease, clinically associated with axial proptosis, marked chemosis, congestion of retinal veins, limitation of extraocular motility, and swelling and pain around the eye. Depending on the severity of the condition, vital structures within the orbit such as the globe and the optic nerve may be compressed or infiltrated by the infection. Generally in children, the orbital cellulitis originates from the ethmoid sinus and may limit itself to the subperiosteal site for a while and later spread into orbital soft tissues.
Acute bacterial orbital cellulitis presents a nonspecific histopathology associated with diffuse polymorphonuclear infiltrate, which may on occasion lead to abscess formation
60 (
Table 17.2). Subperiosteal and soft-tissue abscesses may present with decreased vision, disc edema, and increased intraorbital and intraocular pressures.
Orbital abscess is best demonstrated with MRI. Emergent orbital exploration and the drainage of the abscess are indicated.
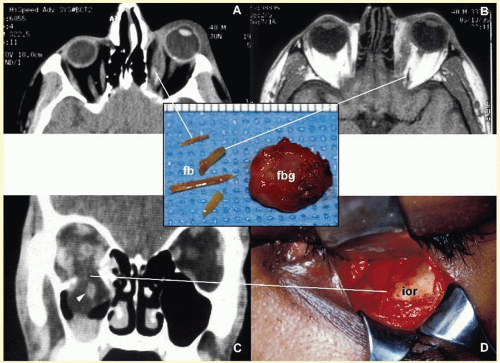
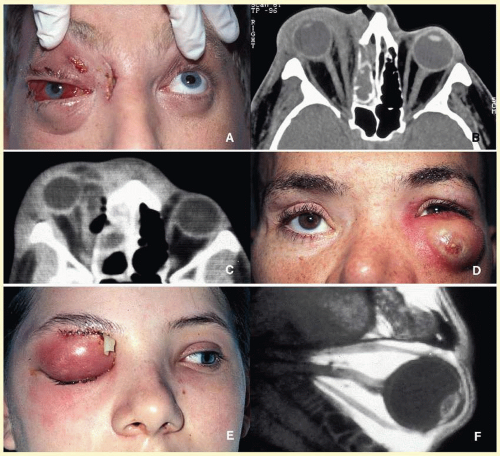
Orbital cellulitis may also be caused by different types of foreign bodies including organic and nonorganic matter and nonautogenous surgical implants.
28 Inorganic foreign materials, such as metal and glass, are usually well tolerated and do not cause infection unless they significantly distort the orbital anatomy with exposure to periorbital sinuses and nasal cavity. Organic foreign bodies such as wood and vegetable fibers, on the other hand, trigger significant foreign body reaction and sometimes suppurative inflammation. These should be documented by CT and/or MRI and surgically removed
29 (
Fig. 17.3). Today many nonautogenous materials are used in ocular and orbital reconstruction, including porous implants, mesh materials, polymeric silicone plates, sponges for scleral buckling procedures, and reservoirs of drainage valves for glaucoma. Implants and repair blocks made of porous materials may lead to acute infection when they erode through a sinus or conjunctival epithelium. Whenever there is a foreign body, noncaseating granulomatous reaction with multinucleated giant cells is identified, adjacent to the foreign material; secondary acute inflammation may be superimposed. In penetrating injuries, the nature of the foreign body is an important factor.
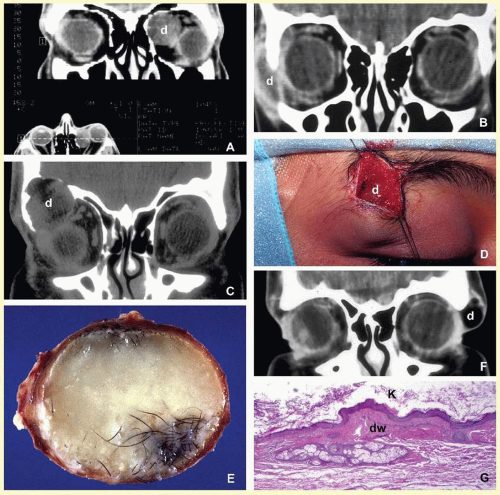
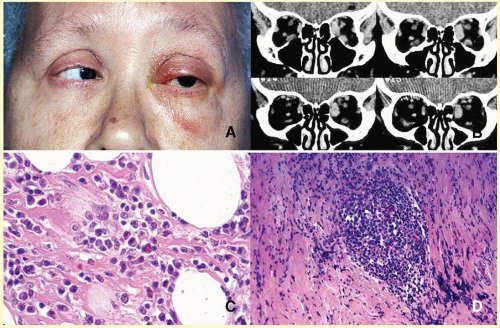
An exception to suppurative soft-tissue reaction caused by bacteria is chronic caseating granulomatous inflammation, which is caused by mycobacteria and certain types of fungi (
Table 17.2). Ocular and adnexal tuberculosis is usually seen with manifestations of systemic mycobacterium infection.
61,62,63 Owing to the recent increase in the numbers of immunologically suppressed individuals secondary to viral epidemics and wider use of immunosuppressant antimetabolites in longer surviving cancer and transplant patients, the incidence of tuberculosis has been rising steadily during the last two decades and the clinical picture of the disease has been changing, with many cases developing due to atypical mycobacteria that are resistant to traditional multidrug treatment.
64 It has been reported that the individuals with HIV/AIDS have an incidence of tuberculosis 500-fold more than that is seen in the general population.
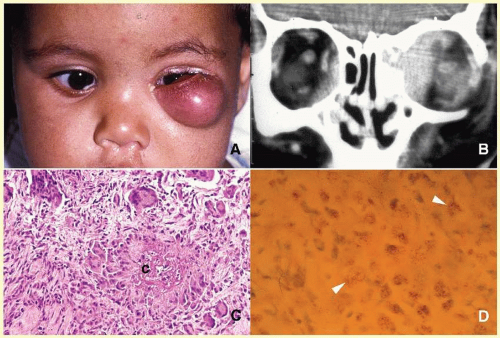
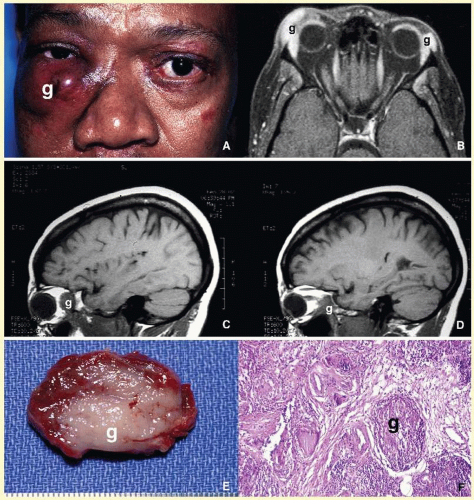
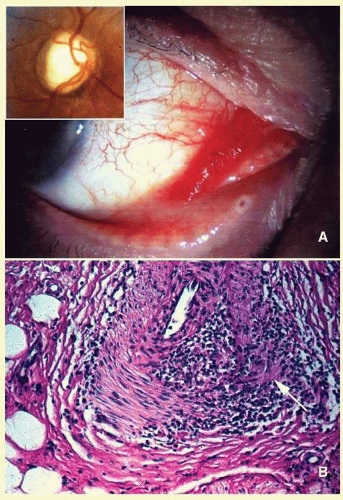
65The orbital disease is more often seen in children and nonwhite patients. History of antecedent penetrating injury is a common presentation of tuberculosis, due to atypical mycobacteria. Histopathology of tuberculosis consists of zonal granulomatous inflammation with numerous epithelioid histiocytes surrounding a necrotic (caseating) center. Tissue diagnosis is pathognomonic only with the documentation of positive acid-fast organisms; however, in many cases, tissue may fail to demonstrate the mycobacteria, but the cultures grow
Mycubacterium tuberculosis or atypical
Mycobacteria. Orbital tuberculosis is usually associated with systemic disease (
Fig. 17.7).
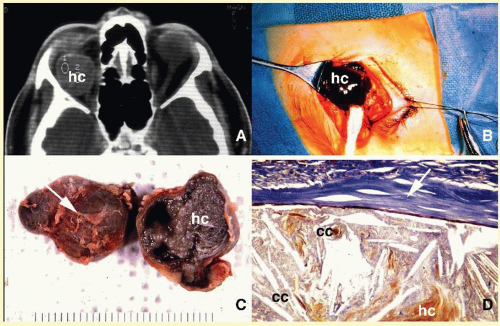
In most instances, fungi infect the orbit as an extension of the paranasal sinus disease or following penetrating injury associated with the introduction of organic matter. Most of the fungal infections, particularly mucormycosis, often develop in immunocompromised patients.
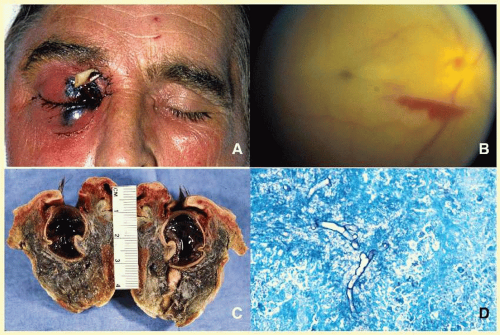
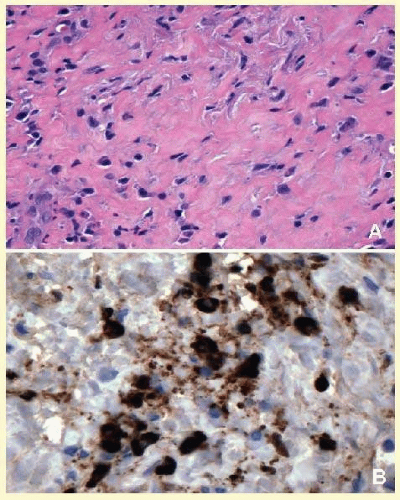
66 Orbital mucormycosis is an emergency situation because it causes rapidly progressing necrotizing inflammation secondary to vascular involvement (
Fig. 17.8). Orbital exploration should be done immediately to establish the diagnosis by identifying the broad, nonseptated hyphae and for surgical debridement as well as irrigation with antifungal agents. The prognosis of mucormycosis is very poor.
Aspergillosis, unlike mucormycosis, presents a low-grade, smoldering chronic granulomatous inflammation, which may be confused with primary orbital tumor.
67 The identification of the organism in fungal and parasitic diseases is crucial at the time of surgery; fungi may also be identified with smears and frozen section.
68 In any kind of orbital exploration secondary to cellulitis, tissue samples should be obtained for gram, fungal, and acid-fast bacillus (AFB) stains and aerobic, anaerobic, and fungal cultures.
As a rule, no matter how rarely, any microbial inflammation, including bacteria, fungi, viruses, and others, may infect the orbit and cause acute, chronic, or granulomatous inflammation leading to tissue damage and fibrosis. These rare diseases include
syphilis,
69 leprosy,
70,71 Lyme disease,
72,73 Parinaud syndrome,
74,75 and
actinomycosis.
76,77 Examples of parasitic orbital infections include
echinococcosis,
cysticercosis,
myiasis, and
trichinosis of extraocular muscles.
78,79,80,81,82,83,84