Fig. 25.1
Diverse causes of the clinically diagnosed “retrocochlear” type of hearing loss. The precise pathology of each case is often difficult to determine clinically. Note that “synaptopathy” refers to a dysfunction of the synapses between hair cells and afferent SGNs, which is audio logically diagnosed as the “retrocochlear” type of hearing loss
25.2 Auditory Neuropathy Spectrum Disorder
Historically, a diagnosis of SGN loss was based on negative recruitment phenomena, positive temporal threshold shift, and poorer speech understanding than that predicted from the pure-tone audiogram threshold. Although these tests retain some importance in the evaluation of retrocochlear hearing loss, they are not applicable to small children and patients with severe cochlear damage. Recently, a group of patients with impaired speech discrimination was found to exhibit a discrepancy between the otoacoustic emission and the auditory brainstem response. These patients are categorized as having an auditory neuropathy spectrum disorder (ANSD). ANSD is defined by (1) absent or atypical auditory brainstem responses and (2) recordable otoacoustic emissions and/or cochlear microphonics, suggesting that the outer hair cells function normally, but the inner hair cells and/or spiral ganglion neurons fail to activate the auditory neural system in the brainstem [5, 6]. The incidence of ANSD is estimated to be 10–15 % in children diagnosed with severe to profound sensorineural hearing loss [7]. Audiometric findings widely vary among patients with ANSD, ranging from congenital profound hearing loss to post-lingual mild hearing loss. In addition to an increase in the pure-tone threshold, ANSD is characterized by speech understanding that is worse than that predicted from the pure-tone audiogram, impaired sound localization, and difficulty in speech perception in a noisy environment [3, 5, 6]. The ANSD is thought to be heterogeneous because the diagnostic criteria for ANSD are not based on the etiology of the disease.
Recent studies have demonstrated a substantial overlap between patients with ANSD and those with cochlear nerve deficiency (CND), especially in the pediatric population [8–11]. As described below, hypoplasia of the bony cochlear nerve canal is often observed in ANSD patients with CND, suggesting that certain types of congenital malformations in the temporal bone are responsible for ANSD in at least part of the pediatric population [11]. Other than congenital malformations of the temporal bone, a considerable number of these cases are caused by dysfunctions in the release of neurotransmitters from the hair cells. Other pathologies include the degeneration of the spiral ganglion cells (ganglionopathy) and demyelinating and/or axonal diseases of the auditory nerve (auditory neuropathy sensu stricto) [3] (Fig. 25.1). These pathological lesions can result in desynchronized auditory nerve activity. In ANSD patients with optic atrophy, a delayed auditory cortical response is detectable despite a negative auditory brainstem response [12]. ANSD patients are now regarded as good candidates for CI [13, 14], but some of these patients experience unsatisfactory results. The severe loss or congenital severe hypoplasia of SGNs is hypothesized in such patients. There have been various attempts to characterize such patients preoperatively.
Imaging studies may supply information about the remaining number of SGNs because the number of SGNs is highly correlated with the diameter of the cochlear nerve [15]. MRI with thin-slice T2-weighted imaging can illustrate the cochlear nerve clearly. ANSD patients with normal cochlear nerves exhibit better speech performance than those with abnormal cochlear nerves [16]. In congenital cases of ANSD, high-resolution CT scans are also useful in the evaluation of anomalies of the cochlear nerve, which is discussed in the following section. Another tool to determine the remaining number of SGNs is the electrophysiological study. ANSD patients with good postoperative speech perception attain significantly higher scores on the electrically evoked auditory brainstem response than those with poor speech perception [16], although the results of electrophysiological studies have not correlated well with those of histological investigations.
25.3 Vestibular Schwannoma
Hearing loss is the most common symptom in vestibular schwannoma patients [17]. Even in patients with preserved hearing, the residual hearing may be lost after treatment, including microsurgery and radiosurgery, or even during careful observation. The pathology of the hearing loss may occur in the cochlea or the auditory nerve, depending on the degrees of compression of the labyrinthine artery and direct damage to the cochlear nerve. Hearing preservation is an unresolved problem in the treatment of vestibular schwannoma patients. Treating neurofibromatosis type II is particularly challenging because these patients may lose bilateral hearing. In such patients, the restoration of hearing is very important. Auditory brainstem implants (ABI) are generally indicated for such patients. Recently, CI was proposed as another option for vestibular schwannoma patients with preserved cochlear nerves. CI surgery is an established and relatively safe procedure, and it can be performed at the time of surgery to remove the vestibular schwannoma via the translabyrinthine approach or after the surgery through the middle cranial fossa or retrosigmoid approach. Unfortunately, however, the performance of CI is widely variable, even among patients with grossly preserved cochlear nerves [18]. This finding indicates that the anatomical preservation of the cochlear nerve does not necessarily indicate its functionality. Damage to the cochlear nerve is unpredictable. The number of SGNs shows only a weak correlation with tumor size, and the pure-tone threshold and speech discrimination scores do not predict the remaining number of SGNs [19].
25.4 Congenital Hearing Loss
Congenital profound sensorineural hearing loss is a main target for CI, and most children with this condition benefit from CIs [20]. In patients with various causes of congenital hearing loss, the average number of SGNs is 14,000–20,000 [21]. This number fulfills the minimum requirement for CI. However, some children have a very small number of SGNs. Histopathological studies have demonstrated a severe loss of SGNs in patients with Cockayne syndrome [22] and xeroderma pigmentosum [23]. These diseases are DNA repair disorders, which can cause severe loss of SGNs [23]. In children with trisomy 13 syndrome [24] and DiGeorge syndrome [21], a total or near-total loss of SGNs have also been reported. These patients are predicted to show a poor outcome after CI, although this has not yet been reported. Children with CHARGE association have traditionally not been regarded as good candidates for CI because the outcome was poor in a considerable proportion of these cases. CHARGE children frequently exhibit inner ear malformations. The typical anomalies include hypoplasia of the cochlea and the absence of the semicircular canals [25, 26]. The histology of CHARGE association is variable. The inner ear may be normal [27], or the modiolus and Rosenthal’s canal may be hypoplastic [28]. This variety may be the reason for the variable outcomes observed after CI in children with CHARGE association [29].
Congenitally deaf children with CND experience poor outcomes after CI. CND is strongly associated with hypoplasia of the bony cochlear nerve canal and narrowing of the internal auditory canal [30]. The fundus of the internal auditory canal forms a pit at the junction with the cochlea. In patients with congenital cochlear nerve deficiency, this structure sometimes becomes narrow and resembles a canal on CT scans. This radiological finding is called a “bony cochlear nerve canal” (Fig. 25.2). A recent study revealed that patients showed poor results after CI when the diameter of the bony cochlear nerve canal was smaller than 1.4 mm [11]. In CHARGE association patients, 77 % of the ears demonstrated cochlear aperture atresia [31]. This finding may be related to radiological findings of a narrow bony cochlear nerve canal, although this has not been demonstrated histologically.
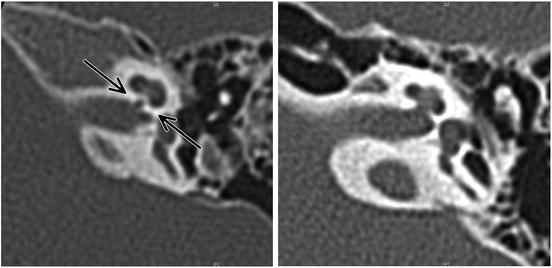
Fig. 25.2
CT images showing narrow (left) and normal (right) bony cochlear nerve canals. Normally, the bony cochlear nerve canal does not resemble a canal. In patients with poorly developed cochlear nerves, however, this structure becomes constricted and resembles a narrow canal (arrow)
The rate of age-related SGN degeneration is approximately 1,000 per decade in individuals with a normal organ of Corti [1], but the degeneration is accelerated in patients with hair cell loss due to a lack of tropic support. A survey of hearing loss patients with various etiologies revealed a rate of SGN degeneration of approximately 2,100 cells per decade [32]. CIs are very effective in congenital SNHL children during early stages of life, but the long-term results remain unknown. It is possible that the effectiveness of CIs decreases later in life due to the loss of SGNs. Intracochlear electrical stimulation suppresses the apoptosis of SGNs, although postmortem histopathological studies have not supported this in human patients. The remaining number of SGNs is smaller in the implanted ear than in the non-implanted ear [33].
25.5 Limitations of Auditory Brainstem Implants in Patients with SGN Degeneration
In patients without an ample number of viable SGNs, auditory brainstem implantation is the only option for the restoration of hearing; however, the results of this procedure have not been satisfactory [34]. Patients with neurofibromatosis type II exhibit particularly poor speech understanding and most of these recipients require visual cues to understand speech. The outcome of auditory brainstem implantation in patients without tumors is superior to that in neurofibromatosis type II patients, but it is inferior to the outcome of CI [35]. In addition, auditory brainstem implants are associated with a risk of electrode migration and malstimulation of the vagus nerve, which can cause lethal complications. Therefore, CI is the preferred auditory implant for patients who may have a sufficient number of SGNs. In patients with a poor performance after CI, an auditory brainstem implant can be regarded as an option. However, the outcome is not promising, as described above. The following chapters discuss attempts to regenerate SGNs, which may improve the performance of CI.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


