Choroideremia
Radha P. Kohly
Rajeev H. Muni
Alan R. Berger
Choroideremia (CHM, OMIM #303100*) is a rare X-linked, hereditary degenerative disease of the choroid, retinal pigment epithelium (RPE), and retina. It is characterized in its early stages by nyctalopia and later by bilateral and symmetric, peripheral visual field constriction. Furthermore, progressive central visual loss occurs in affected males, and more rarely in carrier females. The degree of vision loss is variable among affected individuals, even from the same family, although these differences are greater among members of different families. Nyctalopia typically manifests in early childhood, and peripheral field constriction usually occurs during the second and third decades of life.
Mauthner,1 in1871, first described a disease distinct from retinitis pigmentosa, which he called choroideremia, thinking it represented a congenital absence of the choroid. In 1948, McCulloch and McCulloch2 published the first large report of a Canadian family of Irish origin with choroideremia. This disorder showed X-linked inheritance, and they proposed that choroideremia was therefore distinct from classic retinitis pigmentosa. Choroideremia meets the standard clinical definition of retinitis pigmentosa. However, it is the combination of the underlying genetic cause and the characteristic retinal appearance that make it a distinct entity.
The genetics of choroideremia have been elucidated3 and studies of animal models4 and human mutations now provide insight into the biologic defects underlying this form of night blindness. The pathophysiology of choroideremia, however, is still not well understood mainly owing to the inability to create an animal model that replicates the human condition and the inability to discern the primary site of the disease. The main locus of the disease is a topic of much debate in the literature; some studies suggest the RPE is the primary location, while other studies suggest it is the choriocapillaris or the photoreceptors. Therapeutic research, at this time, is also quite limited partly owing to the lack of an adequate animal model. Gene therapy, however, is promising and may be a potential treatment in the future.
CHOROIDEREMIA-AFFECTED STATUS
Choroideremia is a relentlessly progressive disease that primarily affects males. Night blindness is usually the first symptom to present early in childhood, but it is often appreciated only by the second decade. With progression of the disease, a peripheral ring scotoma develops and gradually expands to involve the far periphery and central vision.
VISION
Central visual acuity is generally maintained until quite late5 in the course of this disease when it usually drops to a level of 20/80 or less. However, there has been a case report of a child as young as 11 years old with almost complete central vision loss.6 Conversely, there have been reports of affected men as old as 65 years of age who have retained normal central acuity.7 Roberts et al8 did a retrospective longitudinal cross-sectional study of the change in visual acuity (VA) in 115 patients with choroideremia over a minimum period of 4.5 years. They found 84% of patients who were younger than 60 years of age had a visual acuity of 20/40 or better and only 33% of patients 60 years of age or older had a VA of 20/200 or worse. The mean 5-year rate of visual acuity change was 0.09 log MAR equivalent. The prognosis for central visual acuity was generally favorable until the seventh decade.
Color vision is usually normal until the later stages of the disease when significant central visual acuity loss occurs.2 There may be a higher incidence of myopia in patients with choroideremia compared to unaffected individuals. The severity of myopia is usually mild but may increase as the choroideremia progresses.9
Early or intermediate stages of choroideremia may rarely be complicated with choroidal neovascular membranes in the fovea10 and may even be seen in carriers who are severely affected with the disease.11 Furthermore, there is a case report of intraretinal neovascularization associated with choroideremia.12 Cataracts are no more frequent in patients with choroideremia than in patients with other types of retinal degenerations.13
OPHTHALMOSCOPIC APPEARANCE
Severity of the disease in males and the manifestation of the carrier females show some interfamilial and intrafamilial variability that can be confusing. Visible fundus changes have been described in patients as young as 1 year of age and virtually total choroidal atrophy described in patients as young as 22 months.2,14 The earliest fundus changes seen in choroideremia are usually seen in childhood and consist of nonspecific mottling and fine granular atrophy of the RPE in the midperiphery or around the peripapillary area (Fig. 25A-1A).15
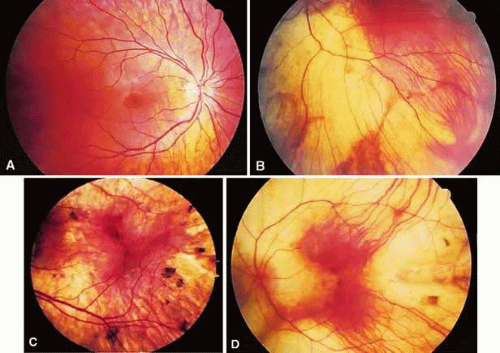 Figure 25A-1. Phenotypic progression of the affected status. A. A 12-year-old boy affected with peripapillary retinal pigment epithelium (RPE) and early choriocapillaris atrophy. B. A 35-year-old man affected with patches of choroidal atrophy. C. A 40-year-old man with a combination of choroidal atrophy and clumping. D. A 45-year-old man affected with severe RPE and choriocapillaris atrophy. |
Over the course of many years, the equatorial choriocapillaris and RPE atrophy progresses centrally and the peripapillary atrophy progresses toward the equator. The progressive loss of the RPE and choriocapillaris results in an exposure of the underlying choroidal vessels that can form confluent scalloped areas where it is often possible to see through to yellow-white sclera. Scattered ribbons of healthy RPE may be seen in the midperiphery between the scalloped areas of atrophy (Fig. 25A-1B). The choroidal vasculature of the macular area and the pigment of the far periphery are usually preserved until the very late stages of the disorder.
In the advanced stages of the disease, widespread chorioretinal atrophy shows the sclera with occasional remnants of the choroidal vasculature in the far periphery, macular area, and around the optic nerve (Fig. 25A-1C). Attenuation of the retinal vessels and optic disc pallor do not generally occur until late in the course of the disease. With time, pigment clumps may be scattered throughout the fundus (Fig. 25A-1D). It is in the advanced stages of the disease where the clinical diagnosis of choroideremia can be confused with other retinal degenerations such as X-linked retinitis pigmentosa, gyrate atrophy, autosomal dominant or recessive diffuse choriocapillaris atrophy, or Wagner vitreoretinal degenration.16
VISUAL FIELDS
Visual field test results usually show good correlation with the clinical fundus appearance, with multiple scotomatous areas corresponding to the retinal disease.17 Ring scotomas and peripheral field constriction usually develop as the disease progresses (Fig. 25A-2). Enlarged blind spots are also a common finding.
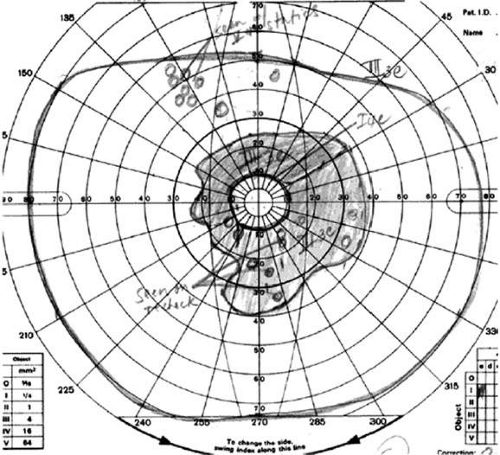 Figure 25A-2. Visual field constriction consistent with advanced peripheral atrophic retinal changes. |
ELECTROPHYSIOLOGY
Electroretinograms (ERGs) may be normal very early in the disease, with the scotopic components of the ERG affected earlier and more severely than the photopic components.15 The absence of a markedly abnormal ERG at an early age, however, does not exclude the diagnosis of choroideremia, especially if the disease is associated with a mutation that results in a milder phenotype.18 Such findings highlight the need for serial ERG testing.18 Sieving et al19 found that rod amplitude responses are generally reduced in affected males with minimally prolonged rod implicit times. Cone amplitudes are initially normal or reduced in amplitudes. Cone implicit times can be delayed even when the cone amplitudes are normal. Mixed rod-cone amplitudes are usually diminished. A negative ERG has also been reported with a b:a wave ratio of 0.5 in an affected male.20,21 Progression of the disease is associated with a steady decline in ERG responses with an amplitude loss of 50% over 4 to 6 years19 that becomes totally extinguished in mid to late adulthood.17 Choroideremia patients generally have abnormal electro-oculographic results and dark adaptation curves (Fig. 25A-3).22,23
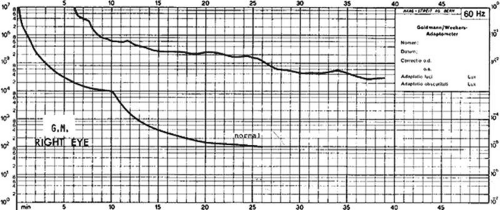 Figure 25A-3. The dark adaptation curve in a young man. Note the absence of scotopic adaptation. (Courtesy of C. McCulloch and S. Arshinoff.) |
Mura et al24 assessed a 4-year-old boy with complete deletion of the choroideremia (CHM) gene by clinical exam, flash ERG, light- and dark-adapted perimetry, and OCT over a period of 3 years. The authors reported that clinically rapid and progressive RPE and choriocapillaris atrophy in the midperiphery and peripapillary areas were found with sparing of the macula. Despite these fundus changes, the authors found that ERG responses were preserved, suggesting that the neurosensory retina was anatomically and functionally less impaired than one might expect based on clinical exam alone. Serial ERGs showed progression first of rod and then of cone disease. OCT findings demonstrated RPE and choriocapillaris atrophy with preservation of the overlying neurosensory retina, although with impaired lamination. Mura et al24 argued that these OCT findings are consistent with the observation of an abnormal fundus appearance in the face of persevered ERG recordings and suggest that RPE changes occur prior to retinal degeneration.
FLUORESCEIN ANGIOGRAPHY
Angiographic changes usually confirm the diagnosis and distinguish choroideremia from classic retinitis pigmentosa. Fluorescein angiography highlights the scalloped edges of choriocapillaris and RPE atrophy that is hypofluorescent next to the hyperfluorescent healthy and patent choriocapillaris (Fig. 25A-4). In the later stages of choroideremia, large choroidal vessels may also degenerate. There are usually isolated areas of normal choriocapillaris in the macular area. Especially in the early stages of the disease, these findings are usually more obvious with fluorescein angiography than with ophthalmoscopy.25,26 Areas of hypofluorescence can also occur by blockage by areas of clumped pigment. Retinal circulation is generally normal, although delayed retinal filling and retinochoroidal anastomoses have been reported.27,28
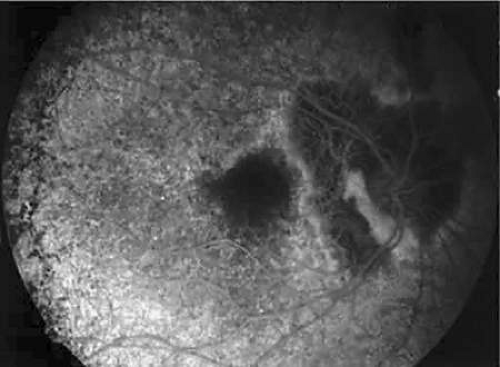 Figure 25A-4. Fluorescein angiography of a 52-year-old woman carrier showing the scalloped edge of the choriocapillaris atrophy. The fundus photograph of this patient is shown in Figure 6B. |
OPTICAL COHERENCE TOMOGRAPHY (OCT)
OCT findings in a 24-year-old male with narrowing of the retinal arterioles, normal-appearing optic nerves, and only an island of intact retina in the center of each macula were documented by Katz et al29 OCT findings demonstrated an abrupt demarcation line between the area of intact retina and the surrounding area of chorioretinal atrophy. Furthermore, the outer retina and RPE were absent in the atrophic areas; however, a thin layer of inner retina and Bruch membrane were intact within these areas. Katz et al29 concluded that the OCT findings were consistent with histopathologic changes previously reported. Also, the authors suggested that the OCT may become a valuable tool in monitoring the preservation of the retina in patients who one day may be treated with gene- or drug-based therapies.
HISTOPATHOLOGY
Histopathologic examination of eyes with choroideremia (Fig. 25A-5) reveals varying degrees of loss of the choroidal vasculature, RPE, photoreceptors, and outer retinal structures. Choroidal and retinal atrophy is often most advanced at the equator, with relative sparing of the macula. McCulloch30 examined the eyes of two elderly males and suggested that choroidal atrophy resulted in secondary loss of the RPE and photoreceptors. Cameron et al31 proposed that the underlying cause of choroideremia is a degeneration in the choroidal vasculature. Syed et al,32 however, suggested that the primary site of disease in choroideremia involved degeneration of the rod photoreceptors. Although there is currently no consensus on which site is initially affected in choroideremia, it does seem clear from most reports that the choroid, RPE, and photoreceptors are all involved in the progression of the disease. The ganglion cells and bipolar cells in the neurosensory retina seem to be spared in choroideremia.33
 Figure 25A-5. Histopathology of the midperiphery of the retina in a male patient with advanced choroideremia. Note the atrophic retina lying against the sclera. (Courtesy of C. McCulloch and S. Arshinoff.) |
Cameron et al31 noted that in patients older than 65 years of age, gliosis of the inner retina and epiretinal membrane formation may be observed in addition to the findings already mentioned. Duplication of the RPE and thickening of Bruch’s membrane has also been reported. Other researchers have reported macrophages in the vicinity of the RPE containing trilaminar curvilinear structures by electron microscopy,31,34 the significance of which is unclear. In addition to the finding of severe degeneration of the choroidal vasculature, RPE, and retina, MacDonald et al33 also found retinal gliosis and mild inflammatory T-cell infiltration surrounding choroidal vessels and in the choroid below transition zones between atrophic and preserved retina. These authors speculated that a reactive inflammatory process at the edges of active disease may be taking place.
GENETIC ASPECTS OF CHOIDEREMIA
It 1942, Goedbloed35 and Waardenburg36 independently discovered that the inheritance of choroideremia was an X-linked recessive trait. In these cases, male-to-male transmission of the disease does not occur because men, by definition, transmit their Y-chromosome to their sons. All daughters of affected men are carriers of the disease and therefore are at 50% risk of transmitting the disease to males (at each conception). Carrier women are also at 50% risk of transmitting the carrier status of choroideremia to female children.
The great variability in fundus appearance found in choroideremia carriers may be explained by the Lyon hypothesis of X-inactivation.37 This theory considers that only one X-chromosome is active in the somatic cells of female carriers. The inactivation of one of the two X-chromosomes occurs randomly early in embryonic life. The inactivated chromosome can be either of paternal or maternal origin. Therefore, a female carrier may develop clones of cells that may express the defective gene as well as clones of normal cells, depending on which X-chromosome is randomly inactivated. In theory, the number of cells with the active X-chromosome inherited from the father should be approximately the same as the number inherited from the mother. In some cases, however, the proportion of active X-chromosomes may be inherited disproportionately from either the father or the mother and may lead to a clinically detectable carrier status or “unfavorable lyonization.” A manifest carrier, owing to “unfavorable lyonization,” is seen when the proportion of X-chromosomes carrying a diseased gene is overly expressed. Favorable lyonization thereby refers to a barely detectable carrier status owing to the small proportion of active X-chromosomes that are carrying the diseased gene. These variable proportions result in cellular mosaicism and account for the great clinical variability observed in funduscopic appearances and ERG testing of choroideremia carriers.
Linkage analysis studies of families affected with choroideremia have allowed the mapping of the gene to the region Xq13-q24.38. The gene for choroideremia, CHM, has been isolated to region Xq21 by positional cloning techniques.38,39
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


