Cataracts and Systemic Disease
Anna K. Junk
Donald A. Morris
Cataracts are frequently found in association with other ocular pathology and may or may not be associated with systemic disease. Lens pathology may evolve secondary to a genetic defect and reveal carrier status as in Lowe syndrome. A distinct appearance of the lens may allow diagnosis of concurrent systemic disease. Exogenous agents, pharmaceuticals, toxins, radiation, infections, and metabolic disorders can cause cataracts and systemic disease. Literature searches were conducted using PubMed.
OCULOCEREBRORENAL SYNDROME OF LOWE
In 1952 Lowe and coauthors1 were the first to recognize congenital cataracts, infantile glaucoma, developmental and mental retardation, and aminoaciduria as a clinical entity. Oculocerebrorenal syndrome of Lowe (OCRL) has been mapped to Xq24-q26 on the X-chromosome.2 Mutations in the OCRL1 gene result in the Lowe syndrome phenotype. The gene encodes for a 105 kD protease, phosphatidylinositol (4,5) bisphosphate (PtdIns [4,5]P2) 5-phosphatase (PIP2 5-phosphatase). PIP2 5-phosphatase, located in the Golgi apparatus, is defective in 1 to 10 males per million in the United States Different mutations in OCRL1 may result in less severe phenotypes with respect to mental retardation and renal dysfunction. Phenotypic variations in OCRL1 mutations range from Dent-2 disease, a renal tubulopathy, to Dent-2 associated with OCRL.3 Deoxyribonucleic acid (DNA) diagnosis and skin fibroblast analysis for PIP2 5-phosphatase is available to confirm OCRL.4
The dense bilateral lens opacities may present as biconvex-shaped cataracts or as posterior, polar, nuclear, or total cataracts. Thinning of the posterior lens capsule and posterior lenticonus has been associated with OCRL. The discoid lens shape may result from loss of lens material through a posterior lens capsule defect5 or due to defective lens fiber formation and subsequent degeneration.6 The cataracts are present at birth, glaucoma may be present congenitally or develop within the first 3 years of life, other ocular findings may include corneal opacity, mitotic pupil, enophthalmos, and hypotonia.7 The cognitive impairment presents with a discrete behavioral phenotype including temper tantrums, irritability, complex repetitive behaviors, and unusual mannerisms. The severe renal Fanconi syndrome may lead to progressive renal impairment. Most boys will develop a distinctive facies, habitus, and attain a height of less than 5 ft due to developmental retardation. Female carriers manifest characteristic but usually asymptomatic lenticular opacities, which will correctly identify carrier status with 100% sensitivity in postpubertal females. They are typically small, irregularly shaped, off-white or gray in color, nonrefractile in appearance, and distributed around the lens equator, more anteriorly than posteriorly (Figs. 41.1 and 41.2). Most importantly and distinctively these opacities are clustered in radial bands or wedges in the peripheral cortex of the lens and visible by retroillumination. Typically, the opacities are moderately dense for 1 or 2 clock-hours, then less numerous or even absent for another clock-hour or 2, and so on. These opacities have to be differentiated from polychromatic, iridescent “crystals” in Steinert myotonic dystrophy, gray-white random opacities in carriers of X-linked adrenoleukodystrophy, sutural opacities in Nance-Horan (NH) syndrome, snowflake granules beneath the anterior and posterior capsule in diabetes mellitus (DM), highly uniform white dots of hypoparathyroidism, and equatorial opacities in cataracta coronaria or ceruleana. Some females also manifest a dense white, central, posterior cortical cataract in the precapsular area. Although the posterior central cataract is apparently congenital, the equatorial and anterior cortical punctate opacities are uncommon in prepubertal female Lowe syndrome carriers.8 Carrier status may also be confirmed by DNA diagnosis.
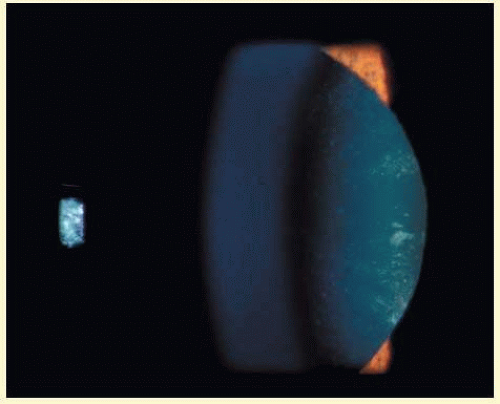 FIG. 41.1 Oculo-cerebro-renal syndrome of Lowe. Female carrier with typical small, irregularly shaped, off-white or gray, nonrefractile opacities in anterior lens cortex, slit lamp view. (Courtesy of Dr. R.A. Lewis, Baylor College, Houston, TX.) |
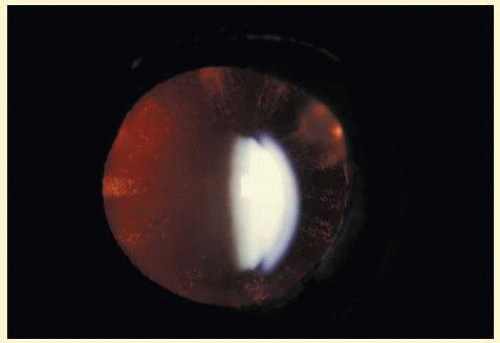 FIG. 41.2 Oculo-cerebro-renal syndrome of Lowe. Retroillumination of small, irregularly shaped, nonrefractile anterior cortical opacities in lens of female carrier of oculo-cerebro-renal syndrome of Lowe. (Courtesy of Dr. R.A. Lewis, Baylor College, Houston, TX.) |
NEUROFIBROMATOSIS 2
Neurofibromatosis 2 (NF2) is an autosomal dominant (AD) disorder that affects 1:25,000 children. It is caused by mutations that inactivate the NF2 tumor suppressor merlin. Multiple CNS and PNS tumors and ocular abnormalities are common in NF2; bilateral vestibular schwannomas (acusticus neurinoma) often result in deafness and are pathognomonic for the disease. Meningeomas, ependynomas, and other cranial nerve and peripheral Schwannomas are also commonly found. The overall survival is estimated to be 38% at 20 years from diagnosis of NF2. Constitutional nonsense or frameshift NF2 mutations are associated with severe NF2 (i.e., earlier onset of symptoms and more tumors), splice site mutations with variable disease severity, and missense mutations with mild disease. Cataracts are the most common nontumor and ocular manifestation in NF2 prevalent in about 60% to 80% NF2 patients. Cultured lens fiber cells lacking NF2 do not fully exit the cell cycle, continue to express epithelial cell markers and lose cell polarity, thus offering an explanation for cataract formation in NF2.9 In one study, the overall prevalence of cataracts in NF2 was 33%, but significantly lower in patients with somatic mosaics and in individuals with new yet unknown mutations and onset of symptoms at ages above 20 years than in patients with classical NF2, nonsense, or frameshift mutations. Of the NF2 patients with cataracts, 29% were diagnosed with cataracts at an age below 10 years and 47% were younger than 20 years. In 70% cataract diagnosis preceded nonocular signs or symptoms of NF2.10 Cataracts typically present as posterior subcapsular plaque-like opacities (Figs. 41.3 and 41.4), other ocular manifestations include retinal hamartomas, optic nerve sheath tumors, fibrotic maculopathies, and perineural calcification of the optic nerve.11
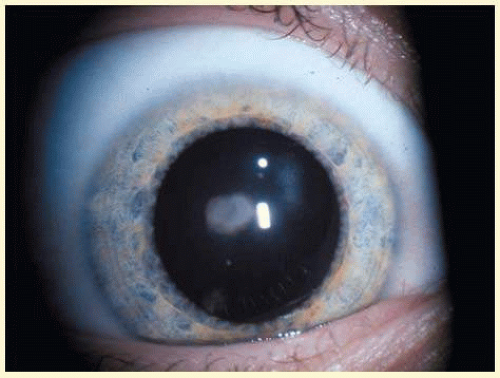 FIG. 41.3 Neurofibromatosis type 2. Paracentral plaque-like retrolental opacity in slit lamp view. (Courtesy of Dr. F.D. Ellis, Zionsville, Indiana.) |
 FIG. 41.4 Neurofibromatosis type 2. Paracentral plaque-like retrolental opacity of NF2 in retroillumination view. (Courtesy of Dr. F.D. Ellis, Zionsville, Indiana.) |
X-LINKED CATARACT NANCE HORAN (NH) SYNDROME
Three types of X-linked cataracts have been reported: congenital total, presenting with posterior sutural opacities in heterozygotes; congenital, with microcornea or slight microphthalmia; and the congenital cataract-dental syndrome or NH syndrome.12 Prenatal detection of congenital bilateral cataract by second trimester ultrasound may lead to the diagnosis of NH syndrome.13 Current linkage analysis is suggestive of allelic heterogeneity in all three types of X-linked cataract. The NH syndrome locus is located within or close to the Xp22.1-Xp22.2 region.14,15 Asymptomatic lens opacities centered around the posterior Y-suture and characteristic facial features such as long face, bulbous nose, and abnormal dentition describe the female carrier phenotype of NH syndrome.16 Affected males present with total congenital cataract, microcornea, dental abnormalities, and dysmorphic features. One-third of affected individuals also present with mental retardation.17 The gene for X-linked isolated congenital cataract has been mapped to Xp22, thus lying within the NH locus, two-thirds of affected patients in one series had complex congenital heart disease.18
LEBER CONGENITAL AMAUROSIS
Leber congenital amaurosis (LCA), prevalent in 3 in 100,000 newborns worldwide, presents with severe vision loss at or near birth. Associated ocular features may include eye poking also known as the oculodigital sign of Franceschetti, ptosis, strabismus, high hyperopia, high myopia, cataracts,19 keratoconus/keratoglobus, microphthalmos, macular coloboma, pigmentary retinopathy and maculopathy, disc edema, and retinal vascular attenuation. Of great interest are the occasional patients with LCA who have a completely normal retinal appearance. A severely reduced or nondetectable electroretinogram (ERG) measured early in the disease process is an absolute diagnostic prerequisite, autosomal recessive (AR) inheritance is commonly found. LCS is variably associated with mental retardation. Six genes have been shown to be mutated in LCA, and they participate in various retinal pathways: retinoid metabolism (RPE65), phototransduction (GUCY2D), photoreceptor outer segment development (CRX), disk morphogenesis (RPGRIP1), zonula adherens formation (CRB1), and cell-cycle progression (AIPL1).20 Longitudinal studies of visual performance show that most LCA patients remain stable, some deteriorate, and rare cases exhibit improvements. Histopathologic analyses reveal that most cases have extensive degenerative retinal changes; some have an entirely normal retinal architecture, whereas others have primitive, poorly developed retinas. RPE65 gene therapy phase I trial results are promising as no systemic toxicity was detected, ocular adverse effects were related to the surgery, and visual function improved in the treated area.21
WILSON DISEASE
Wilson disease is an AR disorder of copper metabolism that manifests in the first or second decade of life in 1:30,000. The abnormal gene (ATP7B) is located on chromosome 13q and encodes a copper-transporting ATPase with two functions: transport of copper into the plasma protein ceruloplasmin and elimination of copper through the bile.22 The hepatocyte’s Golgi apparatus fails to incorporate copper into ceruloplasmin, leading to toxic copper concentrations in the CNS, liver, kidney and eye. Forty percent of patients may present initially with ocular findings. If ocular signs are present, the Kayser-Fleischer corneal ring is most common, found in up to 95% of patients with Wilson disease and eye manifestations. The Kayser-Fleischer ring is a 1- to 3-mm wide golden-brown line located in the deep corneal periphery adjacent to the limbus. Its color is also described as orange-brown, brown-green, golden-yellow, blue, rubyred or a mixture of these colors. Visible copper sulfur deposits in the inner third of Descemet membrane typically begin superiorly, then inferiorly until they complete the Kayser-Fleischer ring, leaving an oval of central clear cornea inside.
Sunflower cataract is named after the petal-shaped alignment of tiny anterior subcapsular copper deposits and is a rare manifestation of Wilson disease (Fig. 41.5), present in 20% to 30% of patients with ocular manifestations. The copper-red to blue-green deposits form a disk-shaped central opacity in the anterior lens capsule usually denser in the pupillary opening with spoke-like radiations toward the periphery. Posterior lens capsule deposits appear in a “fern”-like pattern.23 The lens opacities may reverse on a low-copper diet and systemic d-penicillamine.24
 FIG. 41.5 Sunflower cataract. Copper-red to blue-green deposits in the area of the pupil form a disk-shaped central opacity with patel-shaped spoke-like radiations toward the periphery in patient with Wilson disease. |
Extrapyramidal-motor disturbances of Wilson disease include akinetic-rigidity, choreatic, athetotic and dystonic hyperkinesias. Facial hypomimic, dystonic, and myoclonic symptoms lead to “indifferent expression.” Late stages may paralyze the patient, whereas sensory functions remain preserved. Cerebellar symptoms include nystagmus, blepharospasm, dysarthy, resting, and intentional flapping tremor. Sixty percent of patients develop psychiatric symptoms: appear irritable and aggressive, display diminished intellectual performance, mood changes, depression, and paranoid-hallucinatory syndromes. Hepatosplenomegaly, acute or chronic hepatitis, and cirrhosis complicated by icterus, splenomegaly, esophageal varices, and ascites may develop. Hemolytic anemia, renal osteopathy, and secondary osteomalacia are late manifestations, as is the dark-brown to green discoloration of skin. Other systemic diseases associated with hypercupremia may lead to similar eye findings. Chelating agents such as triethylenetetramine, d-penicillamine, and zinc are the mainstay of therapy. Liver transplants may improve clinical symptoms even if severe neurologic disability is present.
HEREDITARY HYPERFERRITINEMIA-CATARACT SYNDROME
Hereditary hyperferritinemia-cataract syndrome (HHCS) is an AD inherited cataract associated with elevated serum ferritin levels but otherwise normal iron studies and hematologic parameters first described in 1995.25,26 Linkage studies have mapped HHCS to chromosome 19q13, the light chain of ferritin,27 and the major intrinsic protein of lens fiber membrane (MP19).28 The minimum prevalence of HHCS is estimated at 1:200,000.29 To date, 11 different point mutations and 4 small deletions in a highly conserved sequence of an iron-responsive element in the ferritin light chain gene were identified in HHCS families. These mutations lead to the characteristic high-expression levels (10-fold above normal serum levels) of poorly regulated l-ferritin.30 Overexpression and accumulation of l-ferritin in lens fibers results in a highly distinctive, progressive cataract with variability in age of onset and severity.31 Although congenital and infantile cataracts have been reported, HHCS is most commonly diagnosed in childhood and adolescence when patients present with symptoms of glare and severe photophobia exceeding visual loss. The opacities consist of multiple peripheral white, dust-like (pulverulent) dots and larger anterior cortical axial flecks, small crystalline aggregates, and translucent vacuoles best visible by retroillumination. In early stages (neonates and infants), the axial anterior cortex and far periphery were involved first. With progression the midperiphery of the lens will develop smallpunctate opacities, whereas axial cortical aggregates tend to coalesce into radially oriented spokes; few opacities may appear in the nucleus and posterior cortex of the lens. Regular, polyhedral crystalline, diffractive deposits, composed of l-ferritin crystals are seen in light microscopy.32 Pathologic consequences of ferritin accumulation in other tissues have not been reported to date and no systemic treatment is required.
PHENYLKETONURIA
The association of phenylketonuria (PKU) with cataract is controversial.33 PKU is an AR disorder caused by mutations in the gene encoding for phenylalanine hydroxylase, prevalent in 1:4,000 to 1:40,000 newborns. Prenatal and postnatal diagnosis is available and early dietary restriction of phenylalanine in infants and children will avoid severe complications such as mental retardation. A study of 46 untreated patients with PKU detected only 3 in patients (6.5%) with visually significant cataracts. Two of the three patients with untreated PKU and cataracts were examined at the slitlamp. The lens opacities were consistant with prolonged phenothiazine use. Another series detected cataracts in 6 of 11 middle-aged patients with untreated PKU suggesting that the lens opacities may be due to self inflicted trauma or treatment with high dose of thioridazine hydrochloride. Additional findings included phthisis and choroidal hypopigmentation.34 Bilateral lamellar cataracts were reported in one case of PKU.35
MYOTONIC DYSTROPHY
The most common muscular dystrophy in adults, myotonic dystrophy (DM) is an AD disorder based on noncoding microsatellite expansion. As in other disorders caused by microsatellite expansion, there is evidence of anticipation (with each generation disease severity increases and age of onset decreases) and a tendency for maternal transmission of a severe congenital form of the disease. An unstable repeat sequence of triplet nucleotides (CTG) on chromosome 19q13.3 has been identified as the cause in most families (DM1). It results in the CTG expansion at the 3′ untranslated portion of the dystrophia myotonica-protein kinase gene (DMPK). DM2 was mapped to chromosome 3q21 where it causes an untranslated CCTG expansion (mean approximately 5,000 repeats) located in intron 1 of the zinc finger protein 9 (ZNF9) gene.36 Both mutations will result in the typical phenotype of myotonic dystrophy in adults, characterized by hyperexitability of skeletal muscle (myotonia) and muscle degeneration, a conduction defect in cardiac muscle cells, endocrine disorders, and a typical iridescent cataract. The accumulation of these repeat motif expansion RNAs appears to express a preferential affinity to some RNA-binding proteins at the expansion and thus causes a global disruption in RNA splicing and cell metabolism. A third mutation for DM has been postulated but not yet identified.37
Patients with DM2 in general show less anticipation, a milder phenotype, and lack a congenital form. Affected individuals (DM1 and DM2) present with myotony and a characteristic pattern of weakness involving facial, neck flexor, finger flexor, and hip girdle muscles. Severe arrhythmias or progressive cardiomyopathy may be fatal. Hypotestosteronism and oligospermia in males and insulin insensitivity are frequent endocrinologic features. A specific set of serologic changes is present, which includes low γ-globulin, elevated creatine kinase, and elevated follicle-stimulating hormone (FSH) in males. In young adults, dust- and flake-like iridescent and highly refractile multicolored “needles” crisscross the anterior and posterior cortex of the lens (christmas-tree cataract). The colors vary according to the angle of the incident light, and in retroillumination only a dim outline of the cataract is seen (Figs. 41.6 and 41.7). Under the scanning electron microscope, the needles are smooth, rectangular, plate like elements bordered by membranes and amorphous material and running crisscross through the lens. The needles have a high-sulfur content and pronounced S-S, CS-SC, and C-S vibrations in energy-dispersive X-ray and Raman microanalysis.38 They increase and accumulate with age and may eventually result in mature cataract. Rarely, macular and tapeto-retinal pigment epithelial degeneration and optic atrophy are associated with DM.
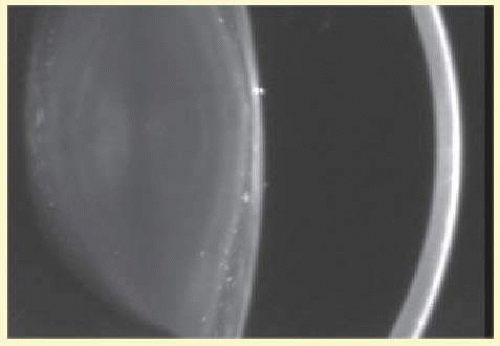 FIG. 41.6 Christmas-tree cataract. Multiple, small irregularly sized opacities in superior and posterior superficial cortex in 43-year-old male patient with myotonic dystrophy (Scheimpflug image). |
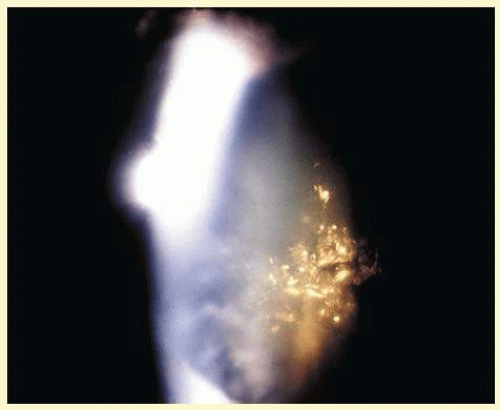 FIG. 41.7 Christmas-tree cataract. Dust- and flake-like iridescent and highly refractile multicolored “needles” crisscross the anterior lens cortex in a patient with myotonic dystrophy. |
Familial mitochondrial myopathy should be differentiated from myotonic dystrophy. Patients present with variable facial weakness, ranging from mild forms to severe progressive ophthalmoplegia associated with facial, pharyngeal, and limb muscle involvement and premature bilateral cataract requiring removal in infancy, adolescence, or early adulthood. Biopsies of skeletal muscles indicate myopathy; histochemistry and electron microscopy reveal abnormal mitochondria in type I fibers. The syndrome is associated with HLA haplotype (A2-B21).39
LYSOSOMAL STORAGE DISORDERS
Most lysosomal storage diseases are associated with deficiencies of glycosidases involved in the catabolism of the sugar chains of glycolipids, oligosaccharides, and glycoproteins. They include single-enzyme deficiencies, the most frequent being Gaucher disease (caused by deficient acid β-glucosidase), Fabry disease (enzyme defect: α-galactosidase A), Tay-Sachs disease (β-hexosaminidase A deficiency), and functional deficiencies of multiple enzymes (reviewed in Kornfeld and Sly40). Less common lysosomal disorders include mucopolysaccharidosis I (MPS I; Hurler/Scheie disease, enzyme defect: α-iduronidase), mucopolysaccharidosis type IVA (Morquio type A; enzyme defect: N-acetylgalactosamine-6-sulfate-sulfatase), GM1-gangliosidosis/mucopolysaccharidosis IVB (Morquio type B; enzyme defect: β-galactosidase), mucolipidosis II/III (enzyme defect: phosphotransferase), mucopolysaccharidosis VI (Maroteaux-Lamy syndrome, enzyme defect: arylsulfatase B), and α-mannosidosis. Extensive clinical heterogeneity is seen in lysosomal storage disorders with regard to the age of onset and severity of symptoms, the organs involved, and CNS effects. Clinically related AR disorders, mucolipidosis, sialidosis, and galactosialidosis (the latter two are both due to deficiency of lysosomal sialidase).41 Although corneal opacities are observed frequently in mucopolysaccharidoses, cataracts have been reported in Hurler syndrome, Morquio (type A and B) syndrome, Fabry disease, and α-mannosidosis. The lysosomal storage disorders associated with cataracts are detailed below.
Mucopolysaccharidosis I (Hurler Syndrome)
MPS I is an AR disorder caused by a deficiency in lysosomal hydrolase, α-l-iduronidase (IDUA), and results in a failure to degrade the glycosaminoglycans dermatan sulfate and heparan sulfate. MPS I presents within a spectrum of clinical phenotypes, where Hurler and Scheie syndromes represent two extremes. The gene encoding IDUA is located on the short arm of chromosome 4, and over 50 mutations have been identified in affected patients. Mutation analysis may help predict a prognosis, but in most cases the distinction between various subtypes is made on clinical grounds. Patients diagnosed at a young age (<18 months) will usually turn out to have a Hurler phenotype, whereas children and adults with Scheie syndrome (milder clinical course and normal life expectancy) are often diagnosed at a later age (>5 years). Intracellular degradation of heparan sulfate and dermatan sulfate are impaired and result in excessive urinary excretion and excess storage. Fibroblasts of MPS I patients with different clinical phenotypes differ in their kinetic parameters of α-iduronidase and glycosaminoglycan accumulation.42 Clinical features of Hurler syndrome, the rapidly progressive severe form of MPS I, include dwarfism, coarse facies, synophrys, saddle nose, hypertrichosis, mental retardation, and deafness, associated with death in the first decade. Diffuse corneal clouding, denser in the limbal periphery than in the corneal center, is the most common ocular finding, but a retinitis pigmentosa (RP)-like picture has also been reported and may be associated with posterior subcapsular cataract. Bone marrow transplantation (BMT) has become a treatment of choice for some patients with Hurler syndrome and has the potential to alter the natural history of the disease.43 The response to this therapy in different organs is variable. The brain can be protected as long as BMT is performed before the onset of irreversible pathology (18 months to 2 years). Hepatosplenomegaly resolves readily, and airway and cardiac muscle functions improve. Cartilage and bone do not respond well to BMT, and patients may require corrective surgery. Cardiac valves will deteriorate with time, corneal clouding never clears completely, and posterior subcapsular cataracts will progress as a consequence of BMT.44 Enzyme replacement therapy for MPS I has been tested in phase I/II and phase III clinical trials and demonstrated effective in patients with attenuated forms of the disease (intermediate/Scheie phenotype).45 The lack of apparent CNS penetration by enzyme currently limits its use in patients with the Hurler phenotype.
MUCOPOLYSACCHARIDOSIS IV (MORQUIO SYNDROME)
The Morquio syndrome is a rare AR mucopolysaccharidosis with a prevalence of 1:76,000. It is characterized by a reduced activity of N-acetylgalactosamine-6-sulfate-sulfatase (type A), or β-galactosidase (type B). The deficiency of β-galactosidase is also responsible for GM1-gangliosisdosis, the severe form of which will lead to fetal hydrops. Fibroblast cultures of a skin biopsy will differentiate Morquio type A and type B, showing reduced activity of N-acetyl-galactosamine-6-sulfate-sulfatase or β-galactosidase, respectively.46 Clinically, both enzyme defects lead to a lysosomal storage disease with an accumulation of keratan sulfate and chondroitin-6-sulfate in connective tissue, skeletal system and teeth. The CNS is not involved. Consequently, abnormalities of the skeletal system (dwarfism), aortic valve disease, and dental abnormalities occur. Ophthalmologically, uniformly distributed diffuse corneal opacification is recognized in early childhood. Only in adulthood, the corneal changes may become clinically significant. Alterations of the trabecular meshwork that may lead to glaucoma can be found.47 Lens opacities have been reported.48 Morquio syndrome may rarely be associated with optic atrophy and tapeto-retinal degeneration.49
Fabry Disease
Fabry disease (angiokeratoma corporis diffusum) is due to any of multiple mutations in the X-linked α-galactosidase A gene with a prevalence of 1:40,000 and an average age at diagnosis between 25 and 30 years. The mutations are kindred-specific, often spontaneous and result in varying degrees of functional enzyme deficiency leading to glycosphingolipid deposits, especially in vascular and reticuloendothelial tissue. Symptoms of anhydrosis, acroparesthesias, rash and renal disease are suggestive of Fabry disease. A recent series of 67 patients50 reports acroparesthesia described as burning or painful “pins and needles” in 100% males (homozygotes) or numbness and increased sensitivity in 53% females (heterozygotes) as the most constant clinical feature. The total or partial inability to sweat, often culminating in “overheating” and precipitation of acroparesthesia, is present in 93% of male patients versus 11% of females. Gastrointestinal symptoms, most commonly chronic diarrhea, are present in 90% of the affected male patients compared with 11% of females. Cardiovascular symptoms are common and include ischemic heart disease, intermittent claudication, paroxysmal palpitations, atrial fibrillation, and episodic dyspnea. Renal involvement, most commonly present as proteinuria, may require renal transplantation. Ninety-three percent of males and 13% of females develop a characteristic angiokeratomatous rash on lower trunk and upper thighs. Males often exhibit a waxy, pallidsallow complexion, thick eyelids, and a subtle coarsening of facial features. Triangular anterior subcapsular cataract was present in 48% males and 14% females, and posterior subcapsular cataract was detected in 9% of homozygous patients but none of the heterozygous individuals. Conjunctival vascular tortuosity and ampulliform vessel dilatations were the most common ocular sign of Fabry’s, present in 100% of males and 83% of females, followed by cornea verticillata in 96% and 76% of males and females, respectively, and retinal vascular tortuosity in 88% and 21%, respectively.50
Another series of 32 homozygous (male) patients with Fabry disease of a mean age of 37 years reports a best-corrected visual acuity of 20/20 in 75%. In this series, cornea verticillata was diagnosed in 44% of eyes, and a corneal haze in 84%. Corneal haze was considered as a sign of disease progression. Twelve patients had bilateral posterior “Fabry” cataracts, feathery opacities radiating subcapsularly from the posterior pole along the posterior suture lines. In five patients, bilateral anterior subcapsular opacities were present as well. Thirty-seven percent of patients had an enlarged blind spot in perimetry, presumably related to subclinical optic neuropathy.51
Sialidosis Type I (Mucolipidosis I)
Lysosomal sialidase (EC 3.2.1.18) has a dual physiologic function; it participates in intralysosomal catabolism of sialylated glycoconjugates and is involved in cellular immune response. Mutations in the sialidase gene NEU1, located on chromosome 6p21.3, result in AR disorder, sialidosis, which is characterized by the progressive lysosomal storage of sialylated glycopeptides and oligosaccharides. Sialidosis type I is a milder, late-onset, normosomatic form of the disorder. Type I patients develop visual defects, myoclonus syndrome, cherry-red macular spots, ataxia, hyperreflexia, and seizures. The severe early-onset form, sialidosis type II, is also associated with dysostosis multiplex, Hurler-like phenotype, mental retardation, and hepatosplenomegaly.41 Patients typically present in the second decade of life with dysarthria, limb ataxia, and intention myoclonus. Eye examination classically reveals cherry-red spots at the macula. Two cases are reported with additional perinuclear (deep cortical) cataracts or punctate subcapsular cataracts.52
Mannosidosis
α-Mannosidosis is an AR lysosomal storage disease caused by a deficiency of lysosomal α-d-mannosidase. Lysosomal α-d-mannosidase is involved in the catabolism of glycoproteins through the sequential degradation of mannose and complex oligosaccharides.53 Clinically, patients present with hepatosplenomegaly, mental retardation, and mild dysostosis and must be differentiated from Hurler-Scheie syndrome. Patients may rarely present with scattered punctate opacities in the entire lens54 or posterior spoke-like cataracts.55 Conjunctival biopsy may confirm the diagnosis if fibroblasts and endothelial cells contain membrane-bound vacuoles and vesicles comprised of homogeneous osmiophilic globules.
SYNDROMES ASSOCIATED WITH ALTERED LENS GEOMETRY
Alport Syndrome
Alport syndrome (AS) is a genetically heterogenous disorder arising from mutations in genes coding for basement membrane type IV collagen. Clinically, the syndrome is characterized by progressive nephropathy, sensorineural hearing loss, and ocular abnormalities, including anterior lenticonus and posterior subcapsular cataract. AS is associated with X-linked (Xq22, 80% to 85% of cases), AR (about 15% of cases), and rarely AD (chromosome 2q) inheritance. Autosomal forms have been attributed to defects in the COL4A3 or COL4A4 genes. The X-linked mutations have been mapped to defects in the α-5-chain of type IV collagen (COL4A5) gene compromising both COL4A3 and COL4A4.56 A subtype of X-linked Alport syndrome (XLAS) in which diffuse leiomyomatosis is an associated feature reflects deletion mutations involving the adjacent COL4A5 and COL4A6 genes. All reported mutations lead to abnormalities in the basement membranes of the glomerulus, cochlea, retina, lens capsule, and cornea, which eventually constitute the typical phenotype.
Anterior lenticonus (Figs. 41.8 and 41.9) is considered an integral part of AS; the anterior capsule thins and allows the lens bulge into the anterior chamber using the pupil as a mold.57 Electron microscopic examination of renal glomeruli and anterior lens capsule confirm capsular thinning and dehiscences.58,59 Anterior lenticonus is typically diagnosed in the second and third decades of life causing clinically significant decreased vision60 but may be present in adolescence and result in spontaneous rupture of the anterior lens capsule.61 Posterior subcapsular cataract is quite frequent, but many patients receive glucocorticosteroids for their renal condition, which may play an etiologic role in these cataracts. Additional ocular features described in XLAS include other corneal dystrophies, microcornea, corneal arcus, iris atrophy, posterior lenticonus, spontaneous lens rupture, spherophakia, a poor macular reflex, fluorescein angiogram hyperfluorescence, electrooculogram and ERG abnormalities, and retinal pigmentation abnormalities.62
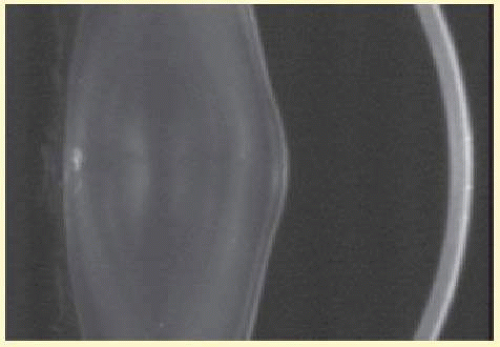 FIG. 41.8 Alport syndrome. Thinned lens capsule and anterior cortex protrude centrally into the anterior chamber and additional small central posterior subcapsular opacity in a 38-year-old male with Alport syndrome (Scheimpflug image). |
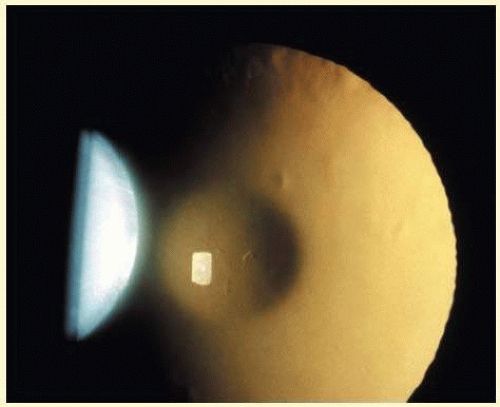 FIG. 41.9 Alport syndrome. Oil-droplet appearance of anterior lenticonus in retroillumination. (Courtesy of Dr. N. Meadow, Manhattan Eye & Ear Hospital, New York, NY.) |
Fechtner Syndrome
Fechtner syndrome is an AD macrothrombocytopenia due to a mutation on chromosome 22 in the MYH9 gene. It encodes for the nonmuscle myosin heavy chain IIA (NMMHC-A),63 which is expressed in platelets, kidney, leukocytes, and cochlea. Fechtner syndrome, also reported as a variant of AS, shares with the latter the triad of nephritis, sensorineural hearing loss, and eye abnormalities and presents the additional features of macrothrombocytopenia and polymorphonuclear inclusions called Döhle-like bodies. The May-Hegglin anomaly, Sebastian syndrome, and Epstein syndrome are macrothrombocytopenias caused by distinct mutations of MYH9 but not associated with cataract-like Fechtner syndrome. They are distinguished from Fechtner syndrome by different combinations of clinical and laboratory signs, considerable overlap exists. A recent report therefore suggested that May-Hegglin anomaly, Sebastian syndrome, Epstein syndrome, and Fechtner syndrome are not distinct entities but rather a single disorder with a continuous clinical spectrum varying from mild macrothrombocytopenia with leukocyte inclusions to a severe form complicated by hearing loss, cataracts, and renal failure. The term “MHY9-related disease” was proposed.64
Posterior Lenticonus
Posterior lenticonus or lentiglobus is a unilateral or bilateral condition caused by asymmetrical thinning and posterior bowing of the posterior lens capsule centrally or peripherally. This has variable effects on the adjacent lens cortex; opacification sometimes occurs, or it may manifest as a high degree of astigmatism that can be irregular but without cataract. More severe cases are associated with a progressive opacity of the lens lamellae in the abnormal area, sometimes with a dense discoid opacity of the posterior pole. Although sporadic cases may exist, many are inherited as an X-linked or AD trait.65 Slit-lamp examination of relatives is therefore important. Posterior lenticonus has been associated with microcornea, Lowe (oculocerebrorenal) syndrome, NH syndrome, Fanconi syndrome, hyperglycinuria, Duane syndrome, and anterior lentiplanus.17,66
CATARACT ASSOCIATED WITH MUTATIONS IN CHOLESTEROL METABOLISM
Inherited defects in enzymes of cholesterol metabolism are associated with cataracts. This association results from the lens’ ongoing requirement for on-site synthesis of cholesterol. Impaired synthesis can lead to alteration of the lens membrane structure. The lens cell membrane contains the highest cholesterol content of any known membrane. Smith-Lemli-Opitz syndrome, cerebrotendinous xanthomatosis (CTX), mevalonic aciduria, and Conradi-Hunermann-Happle (CHH) syndrome all involve mutations in enzymes of cholesterol metabolism and affected patients can develop cataracts.67
Smith-Lemli-Opitz Syndrome (RSH)
Smith-Lemli-Opitz syndrome (SLOS) or RSH (after the initials of the last names of three patients) is an AR disorder of cholesterol biosynthesis with a broad spectrum of phenotypic abnormalities caused by mutations of the 7-dehydrocholesterol reductase gene (DHCR7) on chromosome 11 diagnosed in 1:15,000 to 20,000 live-born newborns.68 Prenatal diagnosis is available by detection of elevated 7-dehydrocholesterol or of SLOS-causing mutations in the DHCR7 gene.69 The clinical phenotype of SLOS includes a lethal form, “idiopathic” hydrops. Neurologic findings include microcephaly with cerebral dysgenesis and demyelinization, agenesis of corpus callosum, and cerebellar vermis dysgenesis. cleft palate, different forms of congenital heart defects, pyloric stenosis, and/or Hirschsprung dysganglionosis, adrenal (cortical) insufficiency, cholestatic liver disease, limb malformations, and genital ambiguity in genetic males. Congenital cataracts are present in 20% of affected patients. Other ophthalmologic signs reported in SLOS include blepharoptosis, epicanthal folds, strabismus, downslanting small palpebral fissures, mild exophthalmos, retinal pigment epithelial defects, pale disks, nystagmus, microcornea, aniridia, postlenticular membrane, Duane retraction syndrome, absence of lacrimal puncta, opsoclonus-like movements, corneal enlargement, abnormal iris insertion, bilateral optic pits, sclerocornea, and optic nerve demyelination.70
Cerebrotendinous Xanthomatosis (Essential Hypercholesterolemic Xanthomatosis)
CTX is an AR neurometabolic disease caused by a deficiency of the mitochondrial sterol 27-hydroxylase. The metabolic defect causes reduced bile acid synthesis and increased plasma and tissue cholestanol levels. Left untreated affected individuals will develop the classical phenotype of juvenile cataracts, neurologic dysfunction, and tendon xanthomas. Diagnosis of CTX prior to neurologic deterioration is crucial to prevent brain damage leading to severe mental and neurologic pathology and death.71 Children with unexplained bilateral cataracts and chronic diarrhea should be worked up to rule out CTX. Treatment with chenodeoxycholic acid will normalize laboratory parameters, stop the diarrhea, and prevent the progression of the disease. After a year of treatment, the motor development delay disappears and the intelligence quotient improves, EEG abnormalities disappear.72 In the natural course of the disease, cataracts be the presenting sign evident between the first and third decades.73 The lens opacities are characterized by small irregular corticonuclear opacities, anterior polar cataracts, and dense posterior subcapsular cataracts.74 Histologically, they contain numerous membranous structures with lipidfilled vacuoles. Bilateral optic atrophy and premature retinal senescence (drusen and retinal pigment epithelial defects) have been reported; the vitreous may contain suspended yellowish flakes resembling cholesterol crystals.75 Palpebral xanthelasmas, corneal lipoid arcus, and proptosis have been reported. Other systemic manifestations include mental retardation, drug-resistant epilepsy, pyramidal and cerebellar signs, brain atrophy, atherosclerosis and myocardial infarction, pulmonary insufficiency, and osteoporosis.
Conradi-Hunermann-Happle Syndrome (X-Chromosomal Dominant Chondrodysplasia Punctata Type II)
The CHH syndrome (X-chromosomal dominant chondrodysplasia punctata type II) is an X-linked dominant disorder that is characterized by ichthyosis, chondrodysplasia punctata, cataracts, and short stature. The disease occurs almost exclusively in females, and it is presumed lethal in males, although a few affected males have been reported. CHH shows increased disease expression in successive generations (anticipation). Diagnosis is confirmed by elevated plasma 8 (9) cholesterol level due to 3-β-hydroxysteroid-delta8, delta7-isomerase deficiency resultant from mutations in the emopamil binding protein gene.76,77
Mevalonic Aciduria (Mevalonate Kinase Deficiency)
Mevalonic aciduria in humans is caused by a genetic deficiency of mevalonate kinase and is characterized by very high-mevalonic acid plasma levels, developmental malformations, and cataracts. Chronic exposure of the lens to mevalonic acid can induce cataracts due to a toxic effect on lens cell membranes, resulting in an apparent progressive increase in membrane permeability.78 In a retrospective review of 50 patients from 38 different families, 60% of patients presented within the first 6 months of life and 92% presented before the age of 5. Systemic symptoms include febrile diarrhea and/or rash in 66% associated with lymphadenopathy, diarrhea, joint pain, skin lesions, abdominal pain, and splenomegaly. In addition to febrile attacks, patients may present with inflammatory bowel disease, erosive polyarthritis, and Sjögren syndrome. The p.Val377Ile mutation was the most commonly identified mutation. Mevalonate kinase deficiency may remain highly active or display decreased disease activity over time.79 Treatment with interleukin 1 antagonists (anakrina) can achieve complete or partial remission.
Disorders Associated with Impaired DNA Repair Mechanisms
DNA repair is a fundamental process designed to keep the integrity of genomic DNA that is continuously challenged by intrinsic or environmentally induced alterations. Failure of DNA repair consequently may affect normal growth and development, aging, programmed cell death (apoptosis), and uncontrolled cell proliferation (cancer). The structure and integrity of DNA may be altered spontaneously because of intrinsic instability of chemical bonds in DNA or endogenously during cell metabolism. External factors such as irradiation, ultraviolet (UV) light, or chemicals predispose affected individuals to skin and eye pathology. The following AR-inherited disorders can be distinguished clinically and in DNA repair assays: xeroderma pigmentosum (XP), Cockayne syndrome (CS), trichothiodystrophy (TTD), Bloom syndrome, Rothmund-Thomson syndrome (RTS), and Werner syndrome.80 Of these, CS, TTD, and Rothmund-Thomson and Werner syndromes are associated with cataract formation and discussed in detail below.
Cockayne Syndrome
CS is a progressive neurologic disorder characterized in infancy by growth failure (cachectic dwarfism), retinal degeneration, and increased sensitivity to sunlight. The incidence of CS is estimated at 1.8 to 2.7 per million livebirth.81 Dependent on the affected gene two clinical phenotypes can be distinguished: type I, “the classical type,” with postnatal onset, and type II, “the severe type,” in which symptoms are present at birth and may lead to death by the age of 6 or 7. The skin manifestations include increased photosensitivity with pealing and scarring and loss of subcutaneous fat. Mental retardation, microcephaly, severe neurologic progressive impairment, sensorineural deafness, ataxia, spasticity, and intracranial calcifications are common neurologic manifestations. Growth failure, “major cachectic dwarfism,” sexual immaturity, and dysmorphic features, such as enophthalmos, beak-like nose, narrow mouth and chin, characterize the phenotype of CS. First described by Cockayne,82 one of the syndrome’s hallmarks is the pigmentary retinal degeneration found in 60% to 100% of patients. “Salt and pepper” type fundus, bone spicules, and optic atrophy have been reported. ERG shows reduced scotopic and photopic responses that appear to correlate to fundus changes and the age of the patient. Cataract, present in 15% to 36% of patients, may be cortical, posterior subcapsular, or nuclear. Cataracts noted at birth or within the first 3 years of life indicate a poor prognosis, as do iris hypoplasia and microphthalmos. Enophthalmos due to lack of orbital fat is common, as are miotic pupils with a poor response to mydriatics. Strabismus, nystagmus, and decreased or absent lacrimation are also associated with the syndrome.83
Laboratory tests for (prenatal) diagnosis are available and will allow for differentiation of inherited DNA repair disorders. Neither the classical DNA repair test nor post-UV radiation unscheduled DNA synthesis, a measure of the nucleotid excision repair pathway, is abnormal in CS. However, the transcription blockage test looking at recovery of RNA synthesis (RRS) 24 hours after UV exposure is abnormal.84 Treatment is symptomatic.
Trichothiodystrophy
Ichthyosis (scaling of the skin), brittle hair and nails (sulfur-deficient proteins), growth, and mental retardation are manifestations of TTD. The incidence of TTD is estimated to be 1.1 to 1.2 per million livebirth.81 Clinical features of TTD are variable although abnormalities are generally noted from birth. Photosensitivity is present in 50% of cases. As in CS, skin cancer is not associated with TTD. Polarizing light microscopy shows a “tiger tail” pattern, caused by a partial or complete absence of the cuticular layer as seen in scanning electron microscopy. Similar to Cockayne patients may display a birdlike face, receding chin, beaked nose, protruding ears, and mild to severe growth retardation. Mental retardation, spasticity, tremor, ataxia, and neurodysmyelination compose the neurologic manifestations of TTD. Immunodeficiency and decreased fertility may be associated. Cataract is the main ophthalmic feature reported, mostly as small punctate opacities present throughout the lens, rarely as zonular cataracts.85 Decreased lacrimation, pigmentary degeneration of the retina, nystagmus, and optic disk atrophy (associated with encephalopathy) are rarely associated. Brittle eyelashes may induce keratitis due to abnormal orientation of lashes. DNA testing on fibroblast cultures shows various results depending on the pathway involved and may not be diagnostic in 50% of cases. Prenatal diagnosis by fetal hair biopsy has been reported. Three genes have been identified to cause TTD, the yet uncloned TTD-A,86 TTD-B, a rare mutation in the XP-B gene, and TTD-D, caused by mutations on XP-D gene.87 The location of a mutation in the XP-B or XP-D gene appeared to determine the clinical phenotype.88 Recently two patients with clinical symptoms of both XP and TTD have been reported.89
Rothmund-Thomson Syndrome
The prevalence of RTS is unknown, todate around 300 cases have been reported in the literature. RTS is transmitted as a genetically heterogenous, AR trait. Two clinical subforms of RTS have been defined: RTSI characterized by poikiloderma, ectodermal dysplasia, and juvenile cataracts, and RTSII characterized by poikiloderma, congenital bone defects, and an increased risk of osteosarcoma in childhood and skin cancer later in life.90 Skin manifestations begin as large, ill-defined areas of erythema occurring shortly after birth. The erythema leads to atrophy of the skin, with telangiectasias, hyper-and hypopigmentation, aggravated by exposure to light. Most patients show growth retardation, dystrophies of nails and teeth, hypogonadism, and alopecia. Early graying, hair loss, and absence or sparseness of eyelashes and eyebrows are common. Bilateral juvenile cataracts have been reported to be a common feature, 73% in one large series.91 The lens opacities are subcapsular, display rapid onset within 2 to 3 months, most commonly at the age of 2 to 4. In a recent series, however, only 2 patients with cataracts were identified in 41 affected individuals.92 Occasionally bilateral glaucoma, retinal coloboma, and chorioretinal atrophy have been described.93 RTS is caused by mutations in the DNA helicase gene RECQL4.94 Therapy is limited to early detection of osteosarcoma with baseline radiographs after the age of 5 and avoidance of sun exposure.
Werner Syndrome
Premature aging, endocrine disturbances (diabetes, hypogonadism), osteoporosis (progeria of the adult), and a high incidence of sarcomas are hallmarks of Werner syndrome. The syndrome typically manifests after puberty. Arteriosclerosis and osteoporosis are common; myocardial infarction and cancer may lead to early death in fourth to sixth decades. The skin becomes shiny and taut, with atrophy of the underlying subcutaneous tissue. The hair grays often before the patient reaches 20 years of age. Loss of orbital fat leads to deep-set eyes, and the nose becomes beak-shaped. Bilateral cataracts appear at a median age of 30 years.95 Subcapsular, cortical, nuclear, zonular, and punctate lens opacities have been reported. Bullous keratopathy is a common postoperative complication and may necessitate penetrating keratoplasty.96 Werner syndrome is caused by AR mutations in the RECQL2 gene encoding for a DNA helicase with exonuclease activity that participates in carcinogenesis.97,98
DISORDERS WITH DERMATOLOGIC MANIFESTATIONS
Anhidrotic Ectodermal Dysplasia (Christ-Siemens-Touraine Syndrome)
Also known as Christ-Siemens-Touraine syndrome, this rare disease presents as a congenital absence of the sweat, sebaceous, and mucous glands, sparse hair, poorly developed teeth, and growth retardation. Ocular manifestations vary from congenital anophthalmos,99 blepharitis, keratopathy, and childhood nuclear cataract100,101 or may be absent. Anhidrotic ectodermal dysplasia manifestations of the lids present with a reduction in or an absence of meibomian glands, dysfunction of the Moll and Zeis glands leading to chronic squamous blepharitis and lacrimal punctal atresia.102 These anomalies may cause severe ocular surface disease developing during the second decade and secondary corneal complications. Inheritance most commonly is X-linked but may be AD or AR. Hypohidrotic/anhidrotic ectodermal dysplasia (HED/EDA) has been ascribed to at least three genes involved in NF-kappaB activation: ectodysplasin (EDA1), EDA-receptor (EDAR), and EDAR-associated death domain (EDARADD). During hair follicle morphogenesis, EDAR is activated by ectodysplasin and uses EDARADD as an adapter to build a signal transducing complex that leads to NF-kappaB activation.103 Two further mutations in the NF-kappaB family are known to cause anhidrotic ectodermal dysplasia with immune deficiency: X-linked recessive anhidrotic ectodermal dysplasia with immunodeficiency is caused by hypomorphic mutations in the gene encoding NF-kappaB essential modulator/I-kappaB kinase (NEMO/IKK-γ), the regulatory subunit of the IKK complex. AD-EDA-ID is caused by a hypermorphic mutation in the gene encoding the inhibitory protein I-kappaBalpha.104
Incontinentia Pigmenti (Bloch-Sulzberger Syndrome)
Incontinentia pigmenti (IP, Bloch-Sulzberger syndrome) was the first genetic disorder to be ascribed to NF-kappaB dysfunction. IP is an X-linked dominant genodermatosis antenatally lethal in males. Females present with abnormal skin pigmentation (100%), dental (90%), skeletal (40%), central nervous (40%), and ocular (16% to 35%) abnormalities. Other dermatologic signs include alopecia and nail dystrophy. Ocular abnormalities consist of proliferative vitreoretinopathy, retinal detachment, strabismus, congenital cataract, microphthalmia, optic nerve atrophy, nystagmus, and iris hypoplasia. A complex rearrangement of the NEMO () gene accounts for 85% of IP patients, and results in undetectable NEMO protein and absent NF-kappaB activation.103,105
Atopic Dermatitis
Atopic dermatitis is a common skin disorder with increasing prevalence. Lead symptoms of this chronic eczematous dermatitis are withered, thickened, scaly skin. AD is often part of the atopic triad including asthma and allergic rhinitis. The pathogenesis of atopic dermatitis has been associated with a T-helper cell type 2-dominant immune phenomenon and has been linked to filaggrin mutations. Five percent to 38% of adolescent and adult patients may develop shield-shaped anterior subcapsular cataracts and posterior subcapsular cataracts that may rapidly develop within weeks during periods of exacerbation of facial skin involvement.106,107 Topical and systemic steroids have been implicated in the development of cataracts and glaucoma in atopic patients.108 Atopic patients are at risk for retinal detachment, presumably due to eye rubbing. Other ocular manifestations include typical signs and symptoms of allergic disease such as prominent folds of the lower lids (Hertoge sign), keratoconjunctivitis, and keratoconus.
CATARACT ASSOCIATED WITH FACIAL DYSMORPHISM
Congenital Cataract Facial Dysmorphism Neuropathy Syndrome
The congenital cataract facial dysmorphism neuropathy (CCFDN) syndrome is a recently delineated AR condition, first described in gypsies in Bulgaria and subsequently detected in Romania, Hungary, and the United States. The CCFDN gene has been linked to a homozygous mutation in the CTDP1 gene on the telomeric region of chromosome 18q, leading to disruption of ribonucleic acid transcription.109 The cardinal features include congenital cataracts and microcornea, dysmorphic facial features hypomyelinating/demyelinating peripheral neuropathy leading to progressive neurologic deficit. Other manifestations include developmental delay, mild intellectual deficit, short stature, skeletal deformities, and hypogonadism. Additional common ophthalmologic findings are micropupil, strabismus, and nystagmus.110
Marinesco-Sjögren Syndrome
The differential diagnosis of CCFDN syndrome includes Marinesco-Sjögren syndrome (MSS), an AR disorder recently mapped to chromosome 5q31,111 where cataracts are reported at a later age and with variable progression.112,113 Cerebellar involvement and chronic myopathy are mandatory for the diagnosis of MSS. Other features that distinguish CCFDN from MSS are the absence of peripheral neuropathy, facial dysmorphism, and microcornea.
Oculomandibulodyscephaly (Hallermann-Streiff-Francois Syndrome)
Hallermann114 and Streiff115 first described patients characterized by “bird face,” congenital cataract, mandibular hypoplasia, and dental abnormalities. The new syndrome was later defined as Hallermann-Streiff syndrome (HSS), underlining the differences with regard to Franceschetti mandibulofacial dysostosis. In HSS patients, the nose appears thin, sharp and hooked; the prominence of the chin is absent in the lateral view; and marked microstomia with aplasia of multiple teeth is present, patients present with short stature, skin atrophy, and hypotrichosis. Ocular symptoms include severe microphthalmos and congenital partial or total cataracts. The cataract if left alone will eventually undergo spontaneous absorption in the second to third year of life, without considerable inflammation.116 Additional ocular involvement may include glaucoma, blue sclera, nystagmus, strabismus, optic nerve colobomas or dysplasia, and chorioretinal atrophy.
HSS shares several clinical characteristics with oculodentodigital dysplasia (ODDD). However, while ODDD is a dominantly inherited disorder due to mutations in the connexin 43 gene GJA1, the inheritance pattern of the HSS syndrome is still debated.117
Craniosynostosis Syndrome (Crouzon Syndrome)
In the AD craniosynostosis syndromes of Crouzon, Pfeiffer, Jackson-Weiss, and Apert, mutations were found in the gene that codes for fibroblast growth factor receptor 2 (FGFR2). Less frequently, mutations are observed in FGFR1 and FGFR3 in some cases of Crouzon and Pfeiffer syndrome. The mutations identified in FGFR2 are located in exons 5 and 7 of the gene that codes for immunoglobulin (Ig)-like chain III and the region linking Ig II and Ig III of the receptor. Identical mutations are found in the clinically distinct syndromes of Crouzon, Pfeiffer, and Jackson-Weiss; furthermore, the same gene defect can result in a highly variable phenotype even within one family. Thus the clinically distinct craniosynostotic syndromes are considered extremes of a spectrum of craniofacial abnormalities and not nosologic entities. A mutation in the homeotic gene MSX2 was the first genetic defect identified in an AD primary craniosynostosis, that is, in craniosynostosis type 2 (Boston type). In Saethre-Chotzen syndrome, the gene coding for transcription factor TWIST is mutated.118
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


