Fig. 19.1
Photomicrograph of the Boston keratoprosthesis type I (left) and II (right) devices
19.2 Indications
The Boston keratoprosthesis type I is indicated for patients with a history of repeated allograft failure, corneal opacity with extensive neovascularization, and in select cases of limbal stem cell deficiency, such as aniridia. Such patients typically have normal eyelids, blink, and tear film. In contrast, patients with severe autoimmune ocular surface diseases, such as Stevens-Johnson syndrome/toxic epidermal necrolysis, mucous membrane pemphigoid, end-stage keratoconjunctivitis sicca, and those after severe chemical burns may be candidates for the type II device [9]. Incomplete eyelid closure and poor quality and quantity of the tear film can lead to detrimental evaporative damage to the corneal tissue carrying the type I keratoprosthesis device. Foreshortening of the conjunctival fornices and symblephara that prevent the retention of a soft contact lens would also endanger the long-term retention of a Boston type I keratoprosthesis [10]. Clearly, end-stage retinal or optic nerve disease and phthisis bulbi are contraindications to keratoprosthesis implantation, regardless of type.
19.3 Techniques
The Boston keratoprosthesis type II device is similar to the type I, except for an additional anterior extension of the optic that allows for implantation through eyelids that have been surgically closed. The optical portion of the device consists of a polymethyl methacrylate (PMMA) front plate, its stem, and a 2 mm anterior nub. The front plate is 5.0 mm in diameter and comes in different dioptric powers. Selection of the appropriate power depends on the patient’s axial length and phakic status [11]. Similar to the type I device, the stem connects the front plate with the back plate. The standard back plate is 8.5 mm in diameter and, in the current iteration, is also made of PMMA. A 7.0 mm PMMA back plate is available for pediatric patients or patients with smaller eyes. A change in the back plate material from PMMA to titanium is currently awaiting approval by the US Food and Drug Administration, since it has been associated with improved clinical outcomes [12]. A corneal graft, preferably a healthy allograft, is implanted between the two plates. The addition of 8 perforations – 1.3 mm each – to the back plate in 1999 reduced rates of keratolysis by facilitating nourishment of the donor corneal stroma with aqueous humor [13, 14]. More recently, an additional row of back plate holes was added for improved exposure of the posterior cornea to aqueous humor. A C-shaped titanium locking ring placed around the posterior stem prevents intraocular disassembly of the keratoprosthesis. During surgery, the device is secured into place by suturing the corneal graft to the host cornea.
19.3.1 Preoperative Evaluation
Identifying the underlying etiology and fully assessing the condition of the ocular surface is paramount in the choice of appropriate candidates for Boston type II keratoprosthesis implantation. Patients with history of Stevens-Johnson syndrome, mucous membrane pemphigoid, end-stage keratoconjunctivitis sicca, or severe chemical burns with evidence of significant symblephara or ankyloblepharon, ocular surface keratinization, and absence of normal blink function and tear production are the best candidates for the type II device.
A detailed history of the ocular condition and previous surgical interventions should be solicited. History of high intraocular pressure and possible glaucomatous damage during the course of the disease is particularly important in predicting final outcome [15–17], especially in cases of severe chemical burns [18, 19]. In severely damaged eyes, ascribing the relative contributions of the cornea, lens, retina, and optic nerve to a greatly reduced visual acuity can be daunting. B-scan ultrasonography is typically necessary to assess the integrity of the posterior segment as well as the presence or absence of an intraocular lens. Tests of central fixation and projection of light are performed using a strong light source. In particular, presence of nasal light projection may predict a positive functional outcome of keratoprosthesis implantation. In the absence of a retinal detachment or particularly dense vitreous debris behind an opaque cornea and lens, loss of nasal projection may be the only finding to suggest end-stage glaucoma. Sometimes, intraocular pressure can be estimated only grossly with digital palpation, since severe corneal pathology may render readings by applanation tonometry or pneumotonometry erroneous. Assessment of the blink mechanism and tear secretion as well as careful slit lamp examination of the anterior segment are invaluable in identifying proper candidates for the type II device. The presence of decreased and incomplete blink rate, lagophthalmos and frank chronic exposure, absent tear secretion, chronic conjunctival inflammation, ocular surface keratinization, fornix foreshortening, and symblephara are important factors to note in the patient evaluation for a type II device. Ocular surface inflammation should be minimized prior to surgery, especially in patients with underlying autoimmune inflammatory conditions. This may justify a systemic immunosuppressant, such as mycophenolate mofetil.
A detailed discussion about the risks and benefits of keratoprosthesis implantation as well as the need for life-long follow-up with a qualified cornea specialist is of utmost importance. Compliance with daily medications and with regular follow-up to recognize and treat indolent infection, corneal perforation, and worsening glaucomatous damage must be stressed during the preoperative evaluation. Noncompliance can readily compromise the visual benefits from the initial surgery and can result in loss of the keratoprosthesis and/or the eye. Patients should be willing to accept that the cosmetic appearance of their eye will change notably with the type II device and that the use of tinted spectacle lenses may be the only option for acceptable cosmesis. Patients with autoimmune inflammatory conditions, such as Stevens-Johnson syndrome and mucous membrane pemphigoid, must accept the possible need for immunosuppression with systemic agents and regular follow-up with a rheumatologist or uveitis specialist if long-term preservation of the keratoprosthesis is to be achieved.
Prior to surgery, selection of a keratoprosthesis with the correct dioptric power depends on the phakic, aphakic, or pseudophakic status of the patient and on the adjunctive procedures (e.g., lens extraction, placement of a glaucoma valve, pars plana vitrectomy) to be done at the time of surgery. In aphakic and phakic patients, an A-scan measurement of the axial length of the eye is required for proper determination of the keratoprosthesis power. In particular, for phakic patients, lens extraction is mandatory. Implantation of a plano posterior chamber intraocular lens may be performed in select cases [20]. Thus, even if capsular support is compromised and intraocular lens implantation has to be aborted, the power of the keratoprosthesis will be appropriate. Without a pars plana vitrectomy, maintaining a two-chamber eye is essential if anterior placement of a glaucoma drainage device is planned at the time of the keratoprosthesis surgery or in the future. For already pseudophakic patients, the surgeon may decide to leave the intraocular lens in place, and thus a pseudophakic keratoprosthesis of standard power would be chosen. Any residual refractive error can be corrected with glasses postoperatively.
The need for additional procedures at the time of surgery is also determined during the preoperative assessment. For eyes that are naive to intraocular surgery, a more conservative approach may be reasonable. In phakic patients, extraction of the crystalline lens is always indicated. Most eyes that receive a type II keratoprosthesis have had prior intraocular surgeries. We recommend an aggressive approach to the prevention of postoperative glaucoma and retinal detachment. When aggressive management is chosen, involvement of a vitreoretinal and a glaucoma surgeon is necessary. In young patients, for those with prior glaucoma or retinal detachment and those with multiple prior surgeries, we typically perform a total iridectomy, extraction of the phakos or pseudophakos and lens capsule, pars plana vitrectomy, and posterior placement of an Ahmed valve.
It is unclear whether a healthy corneal endothelium is necessary for successful implantation and better long-term outcomes of keratoprosthesis [21]. Pending further data, we recommend the use of fresh donor tissue if available. Frozen donor tissue or the patient’s own excised button, the latter if of normal thickness, can serve as carriers for the keratoprosthesis device if the availability of a freshly harvested corneal donor is limited and/or its cost is too great for the socioeconomic circumstances.
19.3.2 Surgical Technique
The duration of the surgery and the extent of periocular tissue dissection typically mandate general anesthesia in cases of Boston keratoprosthesis type II implantation. This is in contrast to standard corneal transplants or type I device implantation where retrobulbar anesthesia may be adequate. The surgical site is prepped using a 5 % povidone iodine preparation in the operative eye and a 10 % preparation to the surrounding skin.
Preparation of the donor corneal graft for implantation of the type II device is mostly similar to type I surgery. The donor corneal graft should be at least equal or greater in diameter than the keratoprosthesis back plate to be used and always at least 7.0 mm in diameter. A donor graft of less than 7.0 mm in diameter with the standard 5.0 mm front plate will not allow for sufficient donor tissue to suture the keratoprosthesis device to the host corneal rim. Thus, for the standard 8.5 mm in diameter back plate, the donor graft should be at least 8.5 mm in diameter and at least 0.5 mm greater in size than the host cornea trephination diameter. For the 7.0 mm back plate, the donor graft should be at least 7.0 mm in diameter.
After selection of the respective donor and host diameters, the donor graft is prepared by performing an inner 3.0 mm trephination using a skin biopsy punch and an outer trephination with a standard trephine. Whether to perform the inner or outer trephination first depends on individual surgeon preference. The type II keratoprosthesis device is then ready to be assembled and this is done in a similar fashion as for the type I device. The front plate is placed facing down on a double-sided adhesive that is provided by the manufacturer and secures assembly of the device. The fresh donor corneal graft with the 3.0 mm central opening is then slid down the stem of the front plate so that its epithelial side comes in contact with the back surface of the front plate. A small amount of viscoelastic is placed on the endothelial surface of the donor graft to minimize trauma and the back plate is then pushed down the stem using a manufacturer-provided assembly tool. This is followed by placement of the titanium locking ring behind the back plate using the same assembly tool. An audible snap during this step indicates that the components are secured into place. This is confirmed by careful inspection of the device under magnification. The assembled device is then immersed into corneal preservation medium until needed.
Prior to trephination of the patient’s cornea, extensive dissection and removal of all ocular surface epithelium is performed in order to prevent postoperative complications due to epithelial encystment beneath surgically closed eyelids. Symblephara are divided and bulbar, forniceal, and tarsal conjunctival epithelium is removed with sharp dissection. After infiltration of the eyelid margins with 1 % lidocaine with epinephrine, the margins are excised taking care not to leave residual eyelash follicles. The host cornea is marked with the appropriate trephine and the limbal and corneal epithelium peripheral to the marked area are removed with sharp dissection prior to trephination. Additional procedures such as pars plana vitrectomy or implantation of a glaucoma drainage implant are performed at this stage by a vitreoretinal or glaucoma surgeon, respectively, and placement of a temporary (e.g., Eckhardt model) keratoprosthesis for wide-angle visualization of the peripheral vitreous and retina may be necessary as an intermediate step. Host cornea trephination is then performed as in traditional penetrating keratoplasty surgery and the removed tissue is sent for histopathologic examination. An iridoplasty should be performed if corectopia is present and the visual axis is obstructed by iris tissue. Total iridectomy should be considered and is well tolerated as surgical eyelid closure after implantation of the type II device reduces postoperative glare. If the patient is phakic, the crystalline lens must be removed. If the patient is pseudophakic and the intraocular lens is stable, then it may be left into place and the pseudophakic keratoprosthesis is used. The assembled type II keratoprosthesis device is brought to the operating field and sutured into the host corneal rim as in a standard penetrating keratoplasty. We typically secure the assembled device with 12 interrupted 9-0 nylon sutures. Knots are rotated toward the host cornea but need not be buried. Once the first four cardinal sutures are placed, a 2–3 mm corneal shield is placed over the keratoprosthesis optic in order to prevent phototoxicity to the retina.
After implantation of the device and prior to surgical closure of the eyelids, peribulbar vancomycin (25 mg in 0.5 ml), ceftazidime (100 mg in 0.5 ml), and triamcinolone (20 mg in 0.5 ml) are administered, as modified by the patient’s medication allergies. The upper and lower tarsi are approximated with two or three interrupted 6-0 vicryl sutures on either side of the keratoprosthesis, and the eyelid margins are closed with 8-0 nylon mattress sutures over plastic bolsters. With the eye in primary gaze, Vannas scissors are used to create a notch in the upper lid and thus allow the keratoprosthesis nub to protrude through the closed eyelids. Prior to reversal of general anesthesia, a retrobulbar anesthetic may be injected to minimize postoperative discomfort. Antibiotic ointment is placed over the skin closure along with a gentle patch and a Fox shield.
19.3.3 Postoperative Care
We recommend indefinite antibiotic prophylaxis, which starts on the first postoperative day, typically with two antibiotics. Administration of a fourth generation fluoroquinolone four times a day, tapered to twice daily over the next month, is combined with twice daily administration of topical vancomycin 1.4 % (14 mg/ml in benzalkonium chloride preservative), the latter begun within the first postoperative week, and both are continued indefinitely [22]. Since the inclusion of vancomycin in the postoperative treatment regimen, the rate of acute Gram-positive endophthalmitis among keratoprosthesis patients has diminished dramatically [23]. Topical corticosteroids, generally prednisolone acetate 1 %, are also started four times a day on the first postoperative day and are tapered off over the next month. Antibiotic ointment to the eyelid margins is discontinued when the skin sutures and bolsters are removed about 2 weeks after surgery (Fig. 19.2).
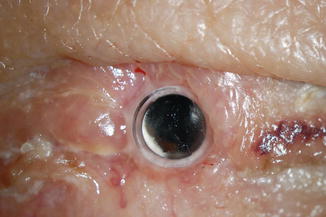
Fig. 19.2
Photomicrograph of the left eye of a patient 2 weeks after Boston keratoprosthesis type II implantation, demonstrating the degree of postoperative eyelid skin closure around the optic of the device
It is important to recognize that once the eyelid skin is fully healed around the keratoprosthesis optic, typically within 2–3 weeks after surgery (Fig. 19.2), topical medications no longer penetrate to the eye. The indefinite administration of a topical fluoroquinolone and vancomycin twice daily aims at reducing microbial colonization of the skin around the keratoprosthesis optic in order to prevent infection reaching the cornea. Moreover, topical glaucoma medications do not penetrate the eye after implantation of the type II device, and any rise in intraocular pressure should be treated with oral acetazolamide or methazolamide.
Follow-up visits are frequent initially in order to assess for postoperative infection, inflammation, and elevation of intraocular pressure. Though follow-up should be individualized, we typically see our patients two to three times in the first two postoperative weeks and then weekly up to the first month after surgery. Examination intervals are then extended to monthly until the first 6 months and then every 2–3 months. If elevation of intraocular pressure is found at any visit, it is prudent to involve a glaucoma specialist in the care of the patient.
19.4 Outcomes
Although numerous studies report outcomes for the Boston keratoprosthesis type I surgery [6, 8, 24–36], there is only a single report on the success rate of Boston keratoprosthesis type II implantation [9] in patients with ocular surface diseases such as mucous membrane pemphigoid, Stevens-Johnson syndrome (Fig. 19.3), and severe chemical burns. In this study from our institution that included 29 eyes that received a type II device over a 10 year period, 50 % of eyes (6 eyes) with mucous membrane pemphigoid and 62.5 % of eyes (5 eyes) with Stevens-Johnson syndrome achieved and maintained a vision of 20/200 or better for more than 2 years. Prior studies [37] had not categorized the eyes by the type of the keratoprosthesis device implanted. Thus, for Yaghouti et al., [37] out of 20 eyes with mucous membrane pemphigoid that received a Boston type I or II keratoprosthesis over an 8-year period (1990–1997) at Massachusetts Eye and Ear Infirmary, 72 % achieved and maintained a visual acuity of at least 20/200 after 2 years and 43 % after 5 years. In the same study, out of 7 eyes with Stevens-Johnson syndrome that received a type I or II keratoprosthesis, only 33 % reached and sustained a vision of at least 20/200 after 2 years and, remarkably, 0 % had retained ≥20/200 acuity 5 years after keratoprosthesis surgery. For patients with Stevens-Johnson syndrome treated over the subsequent 6 years, Sayegh et al. [26] reported that out of 16 eyes, 75 % (12 eyes) had vision equal or better than 20/200 for a mean period of 2.5 ± 2 years after keratoprosthesis (type I or II) implantation.
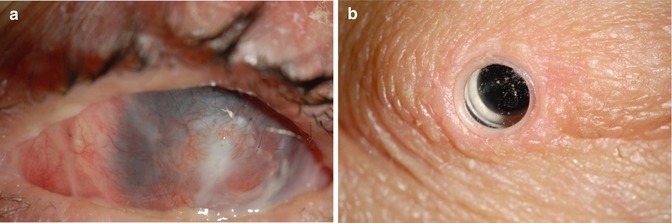
Fig. 19.3
Pre- and postoperative photomicrographs of the left eye of a patient with history of toxic epidermal necrolysis 3 years prior. The patient had undergone keratolimbal allografts, which failed, leading to perforation and necessity for patch graft (a) of the left eye prior to presentation at the Massachusetts Eye and Ear Infirmary. The postoperative (b) vision was 20/20 uncorrected, 3 years postimplantation of a Boston keratoprosthesis type II
In our experience, factors that have improved the outcomes of Boston keratoprosthesis surgery range from an improved design of the device itself to changes in the postoperative care. The transition from a threaded back plate to a snap-in design with a titanium locking ring eliminated intraocular disassembly of the device. The addition of perforations to the back plate reduced the rate of keratolysis by allowing for nourishment of the donor graft with aqueous humor [13, 14]. The change from a PMMA to a titanium back plate, when FDA approved, is predicted to decrease the rate of retroprosthetic membrane formation [12]. Finally, the use of low-dose prophylactic antibiotics indefinitely has diminished the rate of infectious endophthalmitis, at least for the type I device [23]. Due to the small number of type II recipients, it is unclear whether long-term prophylactic antibiotics have a role in preventing infectious complications after a Boston keratoprosthesis type II surgery. In light of the clear advantage of topical antibiotics in the long-term outcomes of the type I device, we also recommend their use for type II recipients. We also tend to favor the implantation of a glaucoma drainage device at the time of keratoprosthesis surgery in most cases, since accurate monitoring of the intraocular pressure is difficult at best after type II surgery, and rapidly progressive glaucoma can quickly and irreversibly compromise vision [9, 15–19, 26, 35, 38–40]. Another adjunctive procedure at the time of keratoprosthesis surgery is pars plana vitrectomy. In theory, release of vitreous traction should decrease the risk of retinal detachment postoperatively, but whether this holds in practice remains to be proven. Finally, whereas it is unclear whether healthy corneal endothelium is necessary for successful implantation and retention of keratoprosthesis [21], we do recommend the use of a healthy graft when possible.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


