Bioavailability
Hari Krishna Ananthula
Ravi D. Vaishya
Megha Barot
Ashim K. Mitra
Bioavailability refers in general to the percentage of a drug that absorbs and reaches the potential site of action regardless of the route of administration. Bioavailability does not have a unique meaning, and a standard acceptable definition does not exist.1 The U.S. Food and Drug Administration defines bioavailability as “the rate and extent to which the active ingredient or active moiety is absorbed from a drug product and becomes available at the site of action. For drug products that are not intended to be absorbed into the bloodstream, bioavailability may be assessed by measurements intended to reflect the rate and extent to which the active ingredient or active moiety becomes available at the site of action.” Since pharmacological response is related to the amount of drug at the site of action, the availability of a drug from a formulation plays a critical role in deciding its clinical efficacy. However, the amount of drug normally cannot be measured directly at the site of action. Hence, most of the pharmacokinetic studies involve the determination of drug concentration in the blood or urine assuming that the drug at the site of action is in equilibrium with the drug in the blood.2
Even though bioavailability is often interpreted as the percentage of the drug absorbed into the bloodstream, the blood levels in topical delivery (such as ophthalmic and dermatologic delivery) are generally measured only to monitor side effects and toxicities and not as an indication of therapeutic efficacy. In addition to the percentage of drug absorbed, bioavailability studies also indicate how the formulation parameters and processes alter both the rate and the consistency of drug absorption. For example, 100% bioavailability does not always suggest that it is therapeutically effective, particularly if the drug absorbs so slowly that it may never reach the minimum therapeutic level. On the other hand, the therapy may elicit toxic response if the drug is absorbed so rapidly that the concentration may exceed the toxic level. Batch-to-batch consistency of absorption coupled with narrow absorption range need to be carefully considered, especially in treating chronic diseases. It is sometimes difficult to discern between disease prognosis and fluctuations in the effectiveness of the medication.
For ocular therapies, both systemic and ocular bioavailability parameters are clinically relevant as both systemic and topical medications are indicated. However, the number of medications given orally to treat ocular infections is considerably less as most of the dosage forms are given either topically or by injections. Some of the medications given intravenously suffer from poor ocular bioavailability and may cause systemic side effects. Ocular drug delivery can be broadly classified into anterior and posterior segment’s delivery. An understanding of the anatomical and physiological aspects of both segments is vital in enhancing the efficacy of ocular drug therapy.
Despite continued investigation of new delivery systems and frequent introduction of novel ophthalmic drugs, drug delivery in the ocular area does not seem to progress at the rapid pace that is typical of other delivery systems, such as oral, transdermal, or transmucosal. Topical ocular delivery normally suffers from low bioavailability and local and systemic side effects. Poor bioavailability of formulations delivered topically to the anterior segment of the eye is due to efficient protective mechanisms that ensure the proper functioning of the eye. The complicating factors include short residence time as a result of rapid and efficient drainage by the nasolacrimal apparatus; noncorneal absorption; and relative impermeability of the cornea to both highly hydrophilic and hydrophobic molecules. Because of these factors, the rate of drug loss from the ocular surface can be 500 to 700 times more rapid than the rate of absorption into the anterior chamber, and 1% to 5% or less of the topically applied dose reaches the anterior segment. Drug delivery to the posterior segment itself is an even bigger challenge along with ocular bioavailability considerations.
An increased understanding of ocular drug absorption mechanisms has spurred researchers to design various ways to enhance ocular bioavailability. Approaches to increase ocular bioavailability and duration of drug action in the anterior segment can be divided into two categories. The first is based on the use of sustained and novel drug delivery systems, such as liposomes, microspheres, nanoparticles, implants, in situ gels, and collagen shields. All these approaches prolong precorneal residence time and provide continuous and controlled drug delivery. The second approach involves optimizing corneal drug absorption/penetration by rational prodrug derivatization and co-administration of therapeutic agents to inhibit efflux pumps. Prodrug design can improve physicochemical properties of the active moiety and has the potential to bypass the efflux pumps. Prodrugs can further be targeted to influx transporters present on the ocular surfaces.
Several other strategies are currently being pursued to improve ocular bioavailability of the existing and conventional ophthalmic dosage forms. These involve optimization of formulation parameters and excipients. Various routes of administrations can also be employed in attaining therapeutic drug concentration in relevant ocular tissues.
This chapter focuses on the opportunities and challenges in ocular drug delivery from a bioavailability point of view. Recent investigations pertaining to ocular drug delivery systems and routes of administration will be discussed.
FACTORS AFFECTING OCULAR BIOAVAILABILITY
Ocular diseases are difficult to treat because of the poor drug bioavailability at the site of action. Apart from the anatomy and physiology of the eye, the other parameters that can limit ocular bioavailability include physicochemical properties of the drug molecule, formulation factors, route of drug delivery, and type of dosage form.
Physiological Factors
Entry of drugs into ocular tissues is restricted because of the eye’s unique anatomy and physiology. Several factors such as precorneal clearance, low corneal permeability, blood-ocular barrier (BOB), and efflux pumps limit the ocular bioavailability.
Precorneal Factors
Topically instilled conventional dosage forms exhibit a very short precorneal residence time and poor ocular bioavailability (<5%) because of the precorneal factors such as tear turnover and drainage, dilution by tear flow, reflex blinking and drug-induced lacrimation. Upon instillation, the drug solution is diluted due to lachrymal secretion and tear flow. Since molecules cross the cornea by simple diffusion, tear dilution lowers the concentration gradient, a driving force for passive absorption of drugs. Therefore, repeated administration is needed to maintain therapeutic drug levels. Tears deliver most of the nutrition and oxygen to the cornea and remove the metabolic waste of avascular corneal epithelium and anterior stroma.3 The tear film is composed of three layers: the innermost mucin film, the middle aqueous layer, and the outer oily layer that retards water evaporation from tear film. The lacrimal gland contributes to the aqueous portion of tear film, goblet cells of conjunctiva provide mucin, and the meibomian gland in the eyelid supplies oil. Blinking action maintains tear film, and nasolacrimal ducts continuously remove the excess tear. Aston et al4 studied permeability of βeta-blockers across the cornea and conjunctiva and found that conjunctival permeability was higher than the corneal permeability. Therefore, It was inferred that tear film, in conjunction with cornea, limits drug permeability.5
With topical instillation, a considerable amount of drug is lost through reflex blinking and excessive tearing. The reasons for excessive tearing may be associated with discomfort or irritation possibly due to components in the formulation, pH of the formulation, or osmolarity difference. When the drop volume is >30 μL (volume that can be accommodated in the cul-de-sac without spillage),6 nasolacrimal drainage and drainage due to gravity are major pathways for drug loss.
Cornea as a Barrier
The cornea is 1 mm thick near limbus and 0.5 mm thick at the center.7 For topically instilled drugs, the corneal route is considered as a major route for the absorption. Steroids, such as dexamethasone, exhibit poor permeability. In an ex vivo transport experiment with excised rabbit corneas, the permeability of dexamethasone was found to be 5E-6 cm per second. Such poor permeability is probably due to the corneal epithelium, which possesses very tight intercellular junctions. However, dexamethasone acetate, which is a prodrug, exhibited a tenfold permeability of 3.7E-5 cm per second.8,9 Molecules cross the cornea by simple diffusion via paracellular and/or transcellular route. Hydrophilic drugs permeate predominantly through intercellular spaces whereas lipophilic drugs diffuse primarily through lipid structures of cells. As a result, hydrophilic molecules prefer the paracellular route and the lipophilic molecules prefer the transcellular route.10 The cornea consists of five different layers, i.e., epithelium, Bowman’s layer, stroma, Descemet’s membrane, and endothelium.7
Corneal epithelium is made up of five to six columnar cell layers and has a thickness of 50 to 100 μm. The paracellular resistance across rabbit cornea is reported to be 12 to 16 kΩ.cm2,11 Presence of the tight junctions in epithelium makes the cornea a formidable barrier for transport of ionized molecules.12 The Bowman’s layer is an acellular layer that divides epithelium and stroma. It is made up of collagen fibers and has a thickness of 8 to 14 μm. The corneal stroma is composed of collagen fibers and provides the most thickness of all the layers. Unlike the Bowman’s layer and the corneal epithelium, stroma is hydrophilic and porous, providing rapid movement of hydrophilic molecules. Nevertheless, for macromolecular and lipophilic drugs, it is a considerable barrier. The Descemet’s membrane is a basement membrane for corneal endothelium that is one cell layer thick (5–10 μm) with phospholipids as a major content. The endothelium is in direct contact with the aqueous humor and controls the movement of molecules from aqueous humor to the stroma. The cells of this layer also have high enzymatic activity. With phospholipids as a major constituent, the endothelium is permeable to lipophilic molecules and nearly impermeable to ions.12,13
In summary, the corneal stroma is hydrophilic and allows rapid movement of hydrophilic molecules and the epithelium is lipophilic and permits ready passage to lipophilic molecules. Overall, the molecules should have good solubility in both oil and water and should also be in nonionized form to achieve optimal corneal absorption.
Blood-Ocular Barrier
The blood-ocular barrier (BOB) is a major barrier to drug entry into the eye after systemic administration. BOB is composed of the blood-aqueous barrier (BAB) and the blood-retinal barrier (BRB). These barriers restrict the entry of xenobiotics and toxins into the eye and protect the globe from inflammation and tissue damage. Overall, BOB controls the movement of nutritional products, metabolic wastes, and exogenous molecules to maintain an environment in which the visual processes can work efficiently. BAB protects the anterior segment. The nonpigmented cell layer of ciliary epithelium and endothelial cells of iridial blood vessels, together constitute the BAB. Both cellular layers express tight intercellular junctions. BAB is not considered a complete barrier like BRB because of the fenestrated capillaries on the ciliary processes. These capillaries are highly permeable and allow ready passage of molecules with molecular weight 40 kDa, which cannot easily pass through iridial blood vessels.14,15 Penetration across an iridial blood vessel from the aqueous humor is less restrictive, and the iris tissue is quite porous. Therefore, drug molecules present in the aqueous humor, especially small and lipophilic ones, can easily enter the circulation and be removed from the anterior chamber.16
BRB is present in the posterior segment and prevents the entry of the drug molecules from blood circulation into the retina. It is further divided into inner BRB and outer BRB. The outer BRB is formed by the monolayer of retinal-pigmented epithelium (RPE), which separates the neural retina from the choroidal blood circulation (choriocapillaries) (Fig. 22-1).17 RPE is firmly attached to the neural retina and plays a major role in maintaining the viability of retinal photoreceptors. In a nutshell, RPE removes waste products of the retina from subretinal space, and responsible for retinoid transport and metabolism, absorption of light, synthesis of growth factors, enzymes and pigments, and immunological factors. It also very selectively allows the passage of nutritional molecules to the retina from choroidal circulation.18
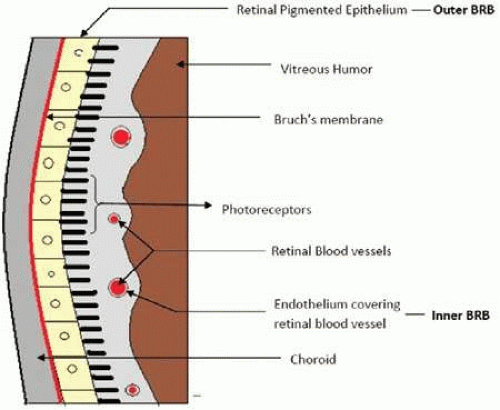 Figure 22.1. Schematic representation of inner BRB and outer BRB. |
The inner BRB is composed of tight junctions between the endothelial cells of the retinal blood vessels (Fig. 22-1). These endothelial cells separate the blood from the neural retina. The tight junctions are made up of proteins such as zonulae occludente.19 The glial cells such as Müller cells and astrocytes also cover the retinal blood vessels and generate signals to stimulate the barrier properties of endothelial cells.20 Anatomically, the inner BRB is quite similar to the blood-brain barrier (BBB). Even though retinal vascular permeability is greater due to higher density of interendothelial junctions and endothelial vesicles, inner BRB has a greater dentisy of tight junctions and pericytes in comparision to BBB. It was suggested that these numerous pericytes act to compensate for a more permeable retinal endothelial barrier.21 The glial cells and the tight junctions of the endothelial cells together limit the entry of drugs into the retina through the paracellular route and protect the neural retina.22
BAB and BRB together play an important role in limiting the entry of xenobiotics from systemic or periocular routes such as oral, subconjunctival, and topical. Apart from acting as a physiological barrier, these ocular structures (especially cornea, inner BRB, and RPE) also express various influx transporters, such as peptide transporter, as well as efflux transporters such as ATP-binding cassette proteins (P-gp and MRP). These transporters can enhance or diminish the drug permeation and can thereby modulate bioavailability.
Routes of Drug Delivery
Four modes of administration can usually be employed for drug delivery to the eye: topical, systemic, intraocular, and periocular (including subconjunctival, sub-Tenon, retrobulbar, and peribulbar).23 Conventional routes (i.e., topical and systemic) of ocular drug delivery and the bioavailability considerations involved are discussed in this section. Other potential routes for local or site-specific delivery such as intraocular and periocular routes will be discussed separately in the following section.
Topical Route
Topical administration is the most common route to deliver ocular medications for treating both surface and intraocular conditions. This route is often preferred over other routes for the management of various pathological diseases affecting the anterior chamber (i.e., cornea, conjunctiva, sclera, anterior uvea). Three main reasons account for this route: (a) convenience for administration, (b) higher ratio of ocular to systemic drug level, and (c) cost affordability. Solutions, emulsions, suspensions, ointments, and rate-control release systems (devices) are commonly used dosage forms administered by the topical route.24 Most of these medications can be easily drained away by the tear flow from the ocular surface, resulting in low bioavailability, which consequently results in very low drug levels in the posterior eye segments (retina and vitreous).16 Therefore, frequent drug administration is necessary to achieve minimum effective concentration even in the anterior segment.25 Corneal and noncorneal are the two specific routes of drug absorption following topical drug administration. Although corneal absorption is the primary pathway for most ophthalmic drugs, it is considered to be a rate-limiting process mainly due to the presence of corneal epithelium.26 As discussed previously, high lipophilicity and presence of tight junctions in the corneal epithelium cells lower the entry of hydrophilic drug molecules into the corneal stroma. However, opening up tight epithelial junctions (e.g., by benzalkonium chloride) is one possible way of increasing hydrophilic drug penetration through the cornea. On the other hand, lipophilic drug substances cross the corneal epithelium much more easily but penetration into the hydrophilic corneal stroma remains somewhat difficult.26 Treatment of superficial infection or inflammation does not require that molecules cross the corneal barrier. However, in most pathological conditions, such as intraocular inflammation and glaucoma, drug penetration into intraocular tissues is essential.27
The endothelium plays a key role in maintaining corneal transparency through its transport and synthetic and secretory functions and is not considered to be a rate-limiting barrier. Leaky junctions of the corneal endothelium result in the passage of macromolecules between the aqueous humor and stroma. The noncorneal path involves penetration across the conjunctiva and sclera into the intraocular tissues. However, it is a minor absorption pathway compared to the corneal route due to the presence of local capillary beds in the conjunctiva and choriocapillaries that remove the drug from target sites into the blood circulation.28 The conjunctival epithelium also constitutes a relatively tight barrier to drug penetration, although it is not as tight as the corneal epithelium. However, the vasculature and lymphatics of the conjunctiva drain a substantial part of the dose that passes the epithelium. The rest that enters the sclera appears to gain access into the retina/choroid. The poorly vascularized sclera is significantly more permeable than the cornea but less permeable than the conjunctiva. In general, the sclera does not form a very tight barrier to penetration of the solutes even of relatively large molecular weight.27,28 Thus, ophthalmic drugs can be absorbed across conjunctiva and delivered to the eye via the sclera. However, drainage loss through blood vessels of the conjunctiva can greatly affect the conjunctival/scleral pathway. Therefore, this noncorneal route is considered to be nonproductive with respect to drug availability in the anterior chamber of the eye. Nevertheless, it should be noted that the conjunctival epithelium is the most viable route for ocular delivery of peptides and oligonucleotides.29
It is much harder to deliver drugs to the posterior segment because of membrane barriers. These barriers, in conjunction with nasolacrimal drainage of administered drops, drug metabolism, protein binding, and long diffusional path lengths result in poor drug delivery to the retina following topical administration.30 Moreover, the passage of drugs from the anterior to the posterior segment does not appear to be an efficient strategy because of the continuous drainage of the aqueous humor (i.e., a turnover of 2.0–3.0 mL/min). Thus, locally used ophthalmic therapies failed to provide an efficient pharmacological effect in the posterior segment (e.g., retina and vitreous).28 Significant efforts have been directed toward enhancing the corneal permeability of drugs following topical administration. The application of high concentrations of penetration enhancer to increase the bioavailability may cause mucosal irritation and corneal abrasion, leading to toxicological complications. In summary, topical application is associated with many complications, such as the blurring of vision, nonproductive absorption, transient precorneal residence time, and instilled solution drainage. As a result, the extent of drug absorption is limited to 5% at best. As ocular barriers and the physicochemical properties of drug molecules govern drug availability in the anterior and posterior segment, research should be directed toward surpassing these barriers by novel routes of administration and/or altering the properties of drug molecules.23
Systemic Route
Drugs administered systemically (e.g., through the oral or intravenous route) also have poor access to the aqueous and vitreous.27 Ocular bioavailability of systemically administered drugs depends on the concentration gradient between serum and ocular tissues and as well as blood-ocular barrier characteristics. Limitations of this route for drug delivery include BOB and systemic toxicity. Barrier characteristics posed by BOB have been discussed earlier in this chapter.
Physicochemical Properties
Transport of drug across anatomical barriers such as the cornea, blood-ocular barrier, and sclera depends on its physicochemical properties: solubility, pKa, partition coefficient (lipophilicity), molecular weight, and molecular size. These properties are discussed in the following sections.
Solubility and pKa
The applied dose has to dissolve in the surrounding medium to cross the biological membrane, except when the formulation is a solution. Solubility is defined as the maximum amount of the solute that can be dissolved in a given solvent under standard conditions of temperature, pressure, and pH. Dissolution and permeation can be two slow rate-limiting steps for absorption. Lipophilic drugs in general have low aqueous solubility, and hence dissolution can be the rate-limiting step. Ionized and hydrophilic molecules permeate poorly through cell membrane owing to poor partitioning into lipid structure (Fig. 22-2). Therefore, permeation can be rate limiting for the hydrophilic or ionized drugs.
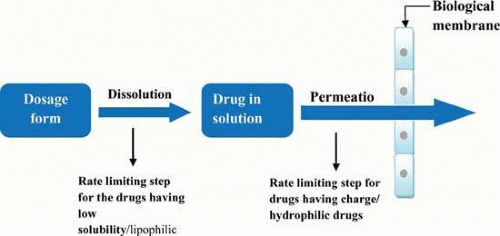 Figure 22.2. Rate-limiting steps for the absorption of drugs at the site of action. |
Aqueous solubility depends on both pH of the solution and pKa of the drug molecule. We know that the acidic drugs having lower pKa are soluble at higher pH. The basic drugs with higher pKa are soluble at lower pH. However, only un-ionized drugs can cross the biological membrane readily and larger amounts of un-ionized drug facilitates absorption. The concentration of the ionized and un-ionized species at a particular pH can be calculated using the Henderson-Hasselbach equation, if the pKa of the drug is known.
For weak acids,
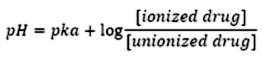
For weak bases,
Tear has a pH of 7.4. At this pH, an acidic drug like penicillin (pKa of 2.7) is highly ionized.
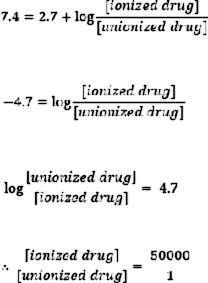
Therefore, one un-ionized molecule exists in the presence of 50,000 ionized molecules. As a result, permeation of penicillin across intact cornea is poor, causing low bioavailability. On the other hand, drugs like sulfamethazine have a pKa of 7.4. Consequently, half of the molecules are in un-ionized form at pH 7.4 and the drug exhibits higher ocular bioavailability.
Therefore, the pKa of the drug and the pH of formulation can affect absorption. As discussed in the previous section, the cornea expresses intercellular tight junctions. Because of these tight junctions, the major route for most of the drugs for transport across the cornea is the transcellular route rather than paracellular. Therefore, the pH of the formulation can affect the corneal penetration of drugs by altering the amount of un-ionized drug. Tear has the pH of approximately 7.4 and is continuously maintained by bicarbonate buffer system, continuous regeneration of tear film, and tear drainage.31 Therefore, the pH of the formulation can be restored to the physiological pH by the precorneal factors. This can affect the corneal penetration. Buffers are used to maintain the pH of formulations within the desirable range so that more drug is available in the non-ionized form. Since the ocular pH is restored to the normal tear pH, the concentration of buffer can have the same effect on the drug permeation. Mitra et al51 reported that increasing the concentration of the citrate buffer (pH was 4.5) from 0 to 0.11M decreased the pharmacological response of pilocarpine significantly (pKa is 6.85). The explanation for this could be that more time will be necessary to restore to normal tear pH as buffer concentration is raised. Homatropine, tropicamide, phenylephrine, and cyclopentolate are some of the drugs having a higher pKa. Hence, these compounds are ionized at the pH of tear and exhibit low corneal permeation. Boberg-Ans et al33 and Wang and Hammarlund34 buffered the pH of tear to 9.2 before administering cyclopentolate, phenylephrine, and homatropine. They observed the same pharmacological response with just one tenth of the normal dose.
Thus, the drug should have a good aqueous solubility to generate a high concentration gradient that allows rapid passive diffusion. It should be present more in the un-ionized form to have appropriate permeability across cellular layers.
Lipophilicity
Partition coefficient is calculated as the ratio of the un-ionized solute concentration in 1-ocatanol and water.
Appreciable aqueous solubility as well as lipophilicity is required for a molecule to permeate optimally through biological membranes. Lipophilicity aids in partitioning of the drug into the lipid membrane. In short, to have a good permeability, a compound should have log P value high enough so that it can easily enter the lipoidal biological membrane. After topical administration, there are two major routes of drug transport to the anterior chamber. One is transcorneal, and other is the conjunctiva-sclera route. The corneal route is a major route for the drug entry into the anterior segment. Most lipophilic molecules can easily permeate through the cornea by the transcorneal route. Nevertheless, the successive layers like stroma are more hydrophilic in nature and preferentially allow permeation of hydrophilic molecules. Phosphate and acetate derivatives of prednisolone and dexamethasone are used topically for ocular inflammation. Both derivatives are easily converted into their parent drug. The basic aim behind making the phosphate prodrug is to increase the water solubility, and that of the acetate prodrug is to increase the lipophilicity, which results in enhanced permeability. Dexamethasone has a log P value of 2.12.35 Its phosphate and acetate derivatives have log P values of 0.54 and 2.92, respectively. As a result of decreased lipophilicity, the permeability of dexamethasone disodium phosphate is 10 times less whereas dexamethasone acetate is 4 times more than that of the parent drug. It has been reported that conjunctival and scleral permeability are apparently not dependant on the partition/distribution coefficient.9
Molecular Weight and Size
Molecular weight and size have a great influence on the bioavailability of drugs as they affect drug permeation across biological barriers. Corneal epithelium has a paracellular pore diameter of 2 nm because of tight junctions and hence it hinders the permeation of drugs with molecular weight >500 Dalton (Da).36 The corneal epithelium is impermeable to the high-molecular weight fluorescein isocyanate-dextran with a molecular weight of 20 kDa.36 Generally, the molecules having a radius up to 4 to 5 Å have been found to have better permeability.9 Stroma has collagen and mucopolysaccharide as major components. Permeability studies done on isolated stroma suggest that the transport across stroma is molecular weight and size dependent.9 The principle route of drug transport across stroma is paracellular. Hence, the intercellular space present between two cells decides what size of molecule will be able to permeate. IgG has a high molecular weight (140 kDa) and molecular radius (50 Å), and hence it exhibits very poor permeation across isolated stroma.37 The endothelium of cornea is not a rate-limiting membrane with respect to molecular weight and molecular size.
Conjunctival membrane is leakier in comparison to cornea or sclera. The paracellular pore size for epithelium is about 3 nm in bulbar conjunctiva and 4.9 nm in the palpebral conjunctiva, which is larger than corneal epithelium. Molecules with a molecular weight of 5 to 10 kDa have a molecular radius in the above range.36 Conjunctival permeability for PEGs (molecular weight range of 238–942 g/mol) is 15 to 25 times more than that of cornea.29 Therefore, conjunctiva allows permeation of small peptides and oligonucleotides.
Sclera is predominantly composed of collagen fibers like cornea. However, the fiber arrangement is completely different. The space between the fibers and cells in sclera is filled with aqueous media, which eases paracellular movement of hydrophilic compounds. Studies clearly show that the scleral permeability depends on the molecular radius and is independent of the distribution coefficient.9
Formulation Factors
A well-designed formulation is mandatory for efficient drug delivery to the target site at therapeutic concentrations. The preferred route for ocular drug delivery is topical, and the formulation is usually a solution or suspension. The other formulations are ointments, ocular inserts, microspheres, and gels. Formulations might contain preservatives, antioxidants, viscosity enhancers, surfactants, penetration enhancers, and mucoadhesives other than the active ingredient. These agents are added to improve the stability of the formulation or to increase the solubility, permeability, and precorneal residence time of the drug. Also, adjustment of osmolarity and dispensed volume can affect the ocular bioavailability.
Instilled Volume
As discussed in the previous sections, <5% of the topically applied drug is bioavailable. The volume of solution that can be instilled in the precorneal pocket is also limited. The eyelid and the conjunctival sac can take up a limited volume of the instilled solution. The precorneal area can accommodate about 30 μL solution. When the instilled volume is >30 μL, the excess solution is removed from the precorneal area via the nasolacrimal duct and the amount of drug in that volume of fluid is lost. From the nasolacrimal duct and nasopharynx, the drug can be absorbed systemically through the nasal mucosa.38,39,40 Moreover, reflex blinking and tearing remove the excess volume. The normal tear volume is restored within 2 to 3 minutes, and the excess volume is lost within 20 to 30 seconds by precorneal factors.6,41 The higher the volume of drop instilled, the more of the applied dose will be lost via nasolacrimal duct system.39,40,42 When the instilled volume is very low, it might be further diluted in the tear fluid, decreasing drug efficacy. For example, 0.5% tropicamide with a drop volume of 5 μL produced less mydriasis than a volume of 16 μL.43
The type of bottle tip or dropper design, dispensing rate, and the angle at which the bottle is held as well as surface tension, viscosity, and temperature can affect the drop volume.44 Usually, dispensing at a 45°-angle produces a smaller drop volume than at the vertical position.38,45 Estacia and Tognan46 studied the effect of administration angle for three prostaglandin formulations. They found that travoprost at 45° angle produced a smaller drop volume than 90° angle. The opposite was true for bimatoprost. Latanoprost showed no significant difference in the drop volume. Therefore, the drop volume should be well studied to avoid drug loss and to generate optimal bioavailability while designing the formulation. In some cases, the instilled volume is lower than the maximum volume. As per Tate’s law, the lower the surface tension of the solution, the smaller will be the size of the dispensed drop. Thus, the agents that can lower the surface tension of fluid can decrease drop volume.38 Commonly, patients use the side press method to dispense the drop, where they apply pressure on the sidewall of the bottle. However, with the bottom press method the pressure is applied on the bottom of the bottle. It is found that the bottom press method produces smaller and more uniform size drops with 25% less force.38 With the increase in the viscosity, the drop volume also increases.47 Temperature can indirectly affect the drop volume by affecting the surface tension of the liquid and the viscosity of the solution.38 Moreover, the dropper design can greatly influence the drop size.
Physiologically, the total human tear volume is approximately 11 μL, with palpebral fissure having approximately 7 μl,1 μl in the precorneal tear film, and approximately 3 μl in each marginal tear meniscus. The palpebral fissure can have approximately 30 μl without overflow under normal physiological conditions, provided that the patient is in an upright position and not blinking.6 Thus the instilled volume should not exceed 30 μL to avoid drug loss.
Viscosity
Lower precorneal retention time has always been a problem for the topically instilled solutions and suspensions. An increase in the viscosity might help to increase the drug retention in the cul-de-sac. Polyvinyl alcohols (PVA), polyvinylpyrrolidone (PVP), hydroxypropyl methylcellulose (HPMC), and hydroxypropyl cellulose (HPC) are some of the polymers that are known to increase the viscosity of the solution.13 The addition of bioadhesive and viscosity-enhancing polymers can help to increase the transcorneal uptake of the drug in the aqueous humor by decreasing the loss of the drug due to tear drainage. Such formulations can prolong the contact time of the drug in the precorneal area, thereby increasing the bioavailability and sustaining the drug release. However, no literature supporting this concept is available. Grass and Robinson48 studied the effect of viscosity on transcorneal permeability of various drugs with different log P values. They found that the drugs having log P in the range of 1 to 4 have no marked increase in the aqueous humor bioavailability even though the viscosity is increased. The reason for this could be their high lipophilicity, which allows the drug to get partitioned quickly into the epithelium, resulting in no significant effect of increased residence time. Increasing viscosity from 1 cps to 90 cps increased the permeability of pilocarpine (log P of 0.22) by 95% and that of pyrilamine (log P of 0.97) by 40%. For drugs having a log P between 1 and 4, there was no significant increase in the permeability. Viscosity of 15 to 30 cps appears to be optimal for ophthalmic solutions.47
Osmolarity
It is necessary to adjust the osmolarity of ophthalmic solutions to that of tears. A solution with equivalent osmolarity to 0.9% NaCl is considered isotonic to tear fluid. If the tonicity of a solution deviates significantly from normal, it may cause discomfort or irritation. This may lead to reflex tearing and blinking, causing drug loss. It is reported that there is no discomfort shown by volunteers for NaCl concentration ranging from 0.5% to 2%.49 However, osmolarity change may lead to reflex blinking and tearing even in the absence of irritation. Moreover, deviation from the isotonicity may cause the cornea to hydrate or dehydrate, which may affect vision.
Penetration Enhancers
As discussed in the previous section, the transcorneal route is a major route for drug absorption or penetration to the anterior chamber. However, the bioavailability of topically instilled drug is usually poor. The reason for this is the barrier property of the cornea imparted by the corneal epithelium and other layers. The noncorneal route, such as the transconjunctival route, also eliminates a considerable amount of topically instilled drug into the systemic circulation. Either an increase in the contact time of an instilled dose with the cornea or transient increase in the corneal permeability can avoid loss of the drug by noncorneal route. The commonly used penetration enhancers are listed in Table 22-1.
Table 22-1 Penetration enhancers used in ophthalmic formulation | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
The penetration enhancers that fall under the category of a surfactant facilitate corneal penetration by changing the barrier properties of the cell membrane and also by solubilization of membrane lipids and proteins. Surfactants are incorporated at low concentrations to transiently alter structure of the cell membrane. However, above a particular concentration known as critical micellar concentration (CMC), the micelles formed can extract membrane phospholipids leading to cell membrane solubilization. Bile salts alter the rheological property of the cell membrane by raising permeability. Their mucolytic action also increases the permeability by causing a temporary change in the membrane structure.13 They can increase the permeation of macromolecules and hydrophilic moieties via porelike structure (paracellular route) rather than the transcellular route.62
The other advantage associated with the use of penetration enhancers is that the drop volume can be lowered due to enhanced drug permeability across the cornea.63 Benzalkonium chloride (BAC) is a well-known preservative having permeation enhancer activity. Higaki64 showed that BAC at a concentration of 0.005% augments the transport of a prostaglandin derivative (S1033) 2.5 times. However, BAC has been reported to be toxic at a concentration of 0.01%, as it disrupted the upper two layers of the cornea and destroyed the intercellular tight junctions.65 Cetylpyridinium chloride (CPC) has also been shown to increase corneal permeability by a similar mechanism.66 Calcium chelators, like EDTA, also increase the permeability of corneal epithelium.4 Chelators bind to available calcium, which ultimately leads to inactivation of protein kinases.67 This effect loosens the tight junction between the corneal epithelial cells and enhances paracellular permeability. Azones can increase the corneal permeability of cyclosporine and produce a better immunesuppression.68 Use of 0.1% azone (laurocapram) increased the corneal absorption of a hydrophilic drug at least 20 times while it impeded the absorption of hydrophobic drug flurbiprofen.57 However, the use of azones at a concentration >0.2% can be irritant and toxic to the eye.58
The penetration enhancers can be valuable adjuvants in an ophthalmic formulation. However, these compounds should be used very cautiously since they can dramatically increase the permeability of cornea. As a result, the enhancers can enter the deeper layers causing damage to the anterior structures. Moreover, BAC and EDTA have been shown to be accumulated in the eye for a longer duration.12 A few penetration enhancers can raise the hydration of corneal tissue. Water-holding capacity of the cornea is because of proteoglycans, which are present between the collagen fibrils. Since cornea is a tissue having low water content, hydration can damage the cell structure. Monti et al studied corneal hydration by various penetration enhancers. BAC and EDTA significantly increased hydration of the cornea while CPC which is also a cationic surfactant similar to BAC, retarded the corneal hydration.60
Surfactants
Surfactants are amphiphilic molecules having both hydrophilic and hydrophobic groups. Surfactants are added to the formulation for various reasons. They can increase the aqueous solubility of drugs. They can act as a preservative since the molecules possess antibacterial activity. These compounds can increase bioavailability by increasing permeation across the cornea. The mechanism behind permeation enhancement is discussed in the previous section. Finally, these surfactants can also increase the wetting property of the vehicle and allow spreading of the drug solution over the corneal surface.
Antioxidants
Oxidation of various active ingredients such as phenylephrine and epinephrine can cause degradation leading to loss of pharmacological activity. Generally, antioxidants are included to avoid oxidative degradation of drugs. Sodium metabisulfite is one of the most widely used antioxidants. Commercially available 10% phenylephrine solution, owing to oxidative degradation, was shown to have the same effectiveness as that of a freshly prepared solution having a strength of 1% to 5%.69 The interaction of drug and oxidant is also an important factor to be considered for the selection of an antioxidant. One good example is epinephrine hydrochloride ophthalmic solution containing sodium bisulfate as an antioxidant. Bisulfate reacts with epinephrine to form adrenochrome sulfonate, which catalyzes the photodegradation of epinephrine.70,71
Dosage Forms
Solutions
Solutions are the most accepted and preferred ophthalmic dosage form. Two main advantages are high patient compliance and self-administration. The drug is available in the solution form and is ready for absorption. However, there are some disadvantages. The drug needs to exhibit high aqueous solubility to produce a high concentration gradient. A significant fraction of drug instilled is lost within 20 to 30 seconds of administration72 owing to precorneal factors. Such short residence time does not allow for drug molecules to partition into lipophilic corneal epithelium. The pKa and lipophilicity of a drug can affect the aqueous and lipid solubility and membrane permeability. Bioavailability can be increased by using penetration enhancers. Cyclodextrins and oligosaccharides have also been used to increase the solubility as well as permeability. These compounds are made of donut-shaped molecules having a hydrophobic inner core and hydrophilic outer surface.73 This hydrophobic core increases the solubility of hydrophobic drugs by complexation inside the core. Cyclodextrins can thus increase the bioavailability and stability while simultaneously lowering tissue irritation.74 Dexamethasone eye drops containing cyclodextrins generate higher bioavailability than simple dexamethasone eye drops and are well tolerated.75 Cyclodextrins have been used to prepare aqueous formulations of various drugs such as ciprofloxacin HCl,76 thalidomide,77 pilocarpine32 and steroids.75 Hydroxypropyl-β-cyclodextrin (HP-β-CD)-based ciprofloxacin HCl formulation performed better than conventional eye drops in terms of stability, bioavailability, and tolerance.76 However, to achieve good bioavailability, cyclodextrins should be used at a suitable concentration (usually <15%) in aqueous formulation.13 At higher concentrations, excess of CD can cause a larger fraction of the complex to remain in the tear solution, causing drug loss through turnover.13 In general, topically applied hydrophilic CD solutions appear to be nontoxic and nonirritating in animals and humans.78 HP-β-CD at a concentration of 12.5% has been shown to be nontoxic to the corneal epithelium45,79 whereas dimethyl-β-CD was found to be toxic within a concentration range of 5% to 12.5% and should be avoided for topical administration.80
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


