Binocular Vision Adaptations and Maldevelopments in Monofixation Syndrome, Microstrabismus, and Macrostrabismus
Binocular Vision Adaptations and Maldevelopments in Monofixation Syndrome, Microstrabismus, and Macrostrabismus
Lawrence Tychsen
Marshall M. Parks
Can we repair defects of visual cortex circuitry in children who have early-onset strabismus? The answer to this question encompasses both sensory and motor behaviors because the clinical hallmarks of the disorder are stereoblindness and absence of motor fusion, which manifests as deviated eyes. Human and animal studies, conducted largely during the last four decades, demonstrate that timely intervention can rescue both sensory and motor fusion. The quality of fusion achieved depends on how early the eyes are realigned and how adaptive the individual’s brain is to rewiring.
MALDEVELOPMENT OF THE VISUAL CORTEX IN UNREPAIRED STRABISMUS
What happens to the brain if eye realignment is delayed considerably or not achieved? What has basic science taught us about the visual cortex in long-duration strabismus? The pioneering work of Hubel and Wiesel1,2,3,4,5 and that of Crawford and von Noorden on animals,6,7,8 decades ago (1977 to 1984), laid important groundwork. If monkeys were made strabismic in infancy and tested at adult age using penetrating electrodes, the majority of neurons within the striate cortex showed deficient binocular responses and reduced disparity sensitivity. These landmark experiments proved that early eye alignment was necessary for development of binocular responsiveness. But several major questions remained unanswered. Were the animals an appropriate behavioral model and did they have the visuomotor deficits of strabismic humans? And what happened structurally within the visual cortex to account for the loss of binocular activity?
The behavioral questions have been answered by studying macaque monkeys that had naturally occurring esotropia. The animals were trained to perform visual fixation and eye-tracking tasks. The experiments documented that the monkeys were a good behavioral model.9,10 They had the full constellation of deficits that serve as clinical markers for early-onset esotropia in humans, including deficits in motor fusion (disparity-driven vergence), latent fixation (fusion maldevelopment) nystagmus, pursuit/optokinetic tracking asymmetries, and asymmetries of motion visually evoked potentials (motion VEPs).11 In normal monkeys, neurons in the superficial layers (layers 2/3) of striate cortex (area V1) are known to provide signals for fusion/disparity sensitivity, whereas neurons in the deeper layers (layer 4B) are important for motion vision and eye tracking.12,13 The behavioral deficits in the strabismic animals implied that they had structural deficits of binocular connections at both superficial and deeper layers of V1.
The structural question was addressed by injecting tracer substances into neurons of area V1 to reveal the underlying circuitry. The tracers are taken up by individual neuronal soma (pyramidal cell bodies) and actively transported within the neurons so as to label their axonal connections. The visual cortex is also processed using labels that reveal metabolic activity.
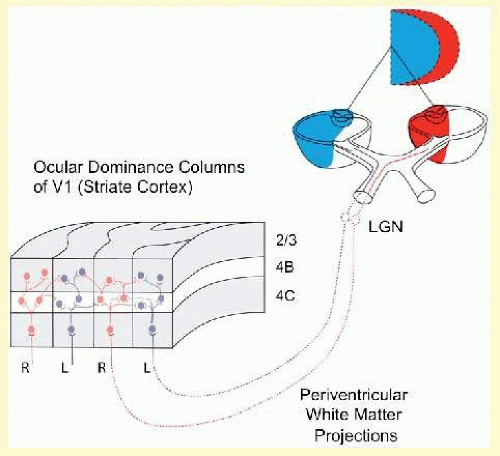
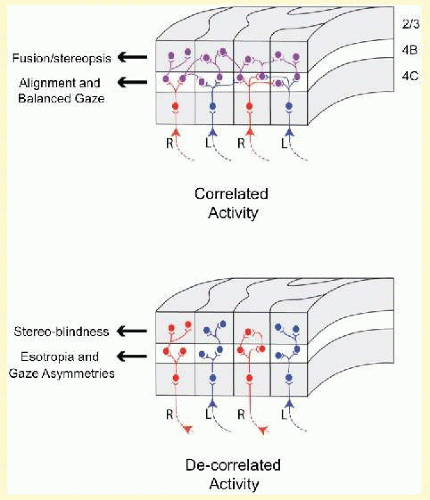
Recall that in normal primates, V1 is organized into columns of neurons (ocular dominance columns [ODCs]) such that every other column is driven exclusively by the right or by the left eye (Fig. 8.1).5 Binocular vision and binocular motor fusion are made possible by horizontal connections between these monocular columns. Binocular connections mediate sharing of information between neighboring ODCs of opposite ocularity, like the horizontal wires interconnecting a row of telephone poles.14,15,16,17 In normal monkeys (Fig. 8.2A), abundant binocular, horizontal, axonal connections join neighboring right- and left-eye columns at the cortical layers known to mediate fusion/stereopsis (layers 2/3) and eye tracking/motion vision (layer 4B).
FIG. 8.1 Neuroanatomic basis for binocular vision. Monocular retinogeniculate projections from left eye (temporal retinanasal visual hemifield) and right eye (nasal retina-temporal hemifield) remain segregated up to and within the input layer of ODCs in V1, layer 4C (striate visual cortex). Binocular vision is made possible by horizontal connections between ODCs of opposite ocularity in upper layers 4B and 2/3 (as well as lower layers 5/6, not shown). RE inputs = red; LE inputs = blue.
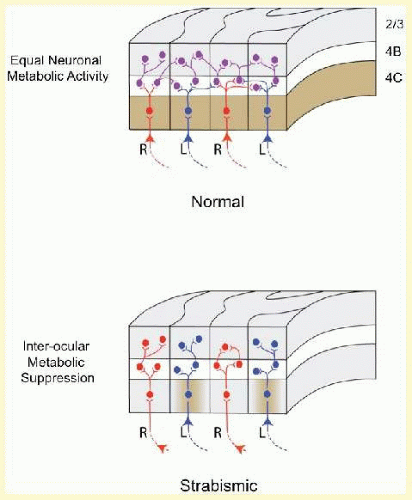
V1 of the monkeys with unrepaired esotropia shows two major abnormalities. The first is an approximately 50% reduction of binocular connections (Fig. 8.2B).18,19 Monocular connections are preserved. Right-eye ODCs connect to other right-eye ODCs, and left-eye ODCs to other left-eye ODCs, but right-eye ODCs make substantially fewer connections to left-eye ODCs. The second abnormality is a striking pattern of “suppression” evident in every other row of columns (Fig. 8.3B), revealed when flattened sections of V1 are processed for cytochrome oxidase20,21,22 (an enzyme within neurons that reveals premortem activity23,24). Misalignment of the eyes causes conflicting (decorrelated) activity in right- and left-eye ODCs, promoting abnormal, inhibitory interactions, mediated presumably by the 50% of connections that remain. The suppression causes down regulation of the neuronal activity within every other row of ODCs.
FIG. 8.2 Horizontal connections for binocular vision in V1 of normal (correlated activity) versus strabismic (decorrelated) primate, layer 2-4B. V1 of normal primates is characterized by equal numbers of monocular and binocular connections. In strabismic primates, the connections are predominantly monocular (i.e., a paucity of binocular connections). RE inputs = red; LE = blue; binocular = violet.
Summarizing these findings, the neuroanatomy of unrepaired strabismus is bad news: chronic metabolic suppression of at least one-half of V1 input neurons and loss of an average of one-half of V1 binocular connections. The good news—implied by the functional recovery after early surgery in humans25,26,27,28,29,30,31—is that the cortex appears to be able to repair these horizontal connections so long as the conflicting activity in neighboring ODCs, caused by image decorrelation, is not allowed to persist longer than a few months. Work from the primate laboratory reinforces this point. When esotropia is repaired in infant monkeys by age 3 weeks (the equivalent of 3 months in human), they regain normal fusion and the full complement of V1 binocular connections.32,33 They also show normal metabolic activity, that is, no suppression. (Marshall Parks preferred that the terms “suppression” and “ARC” not be used in the context of infantile esotropia or monofixation syndrome. For an explanation, see below.)
FIG. 8.3 Metabolic activity in neighboring ODCs within V1 of normal versus strabismic primate. In normal, layer 4C stains uniformly for the metabolic enzyme cytochrome oxidase (shown as brown), indicating equal activity in right-eye versus left-eye columns. In strabismic, a narrow monocular zone within the dominant ODCs (shown here as left eye) shows normal metabolic activity (brown), but ODCs belonging to the suppressed eye (shown as right eye) and binocular border zones between ODCs are pale, connoting abnormally low—that is, suppressed—activity.
An additional clinical implication can be drawn from the metabolic suppression findings. Jampolsky34,35 has long advocated alternating occlusion of strabismic infants up to the date of surgical repair. His argument—dating from 1978—assumes that alternating occlusion abolishes the conflicting (decorrelated) signals that give rise to interocular suppression. The metabolic findings support his recommendation.
VISUAL CORTEX MECHANISMS IN MONOFIXATION SYNDROME
When disparity-driven, fusional vergence is tested in monkeys with large-angle esotropia, a complete absence of motor fusion is evident, similar to what is observed clinically in humans with large-angle esotropia.36 But other strabismic monkeys with angles of heterotropia approximately 4° (8 prism diopter [PD]) show some capacity for fusional vergence.10,36 In other words, their ocular motor behavior resembles that of humans with monofixation syndrome.
The major sensory and motor features of monofixation syndrome are listed in Table 8-1. These features were delineated by Parks37,38 in his 1969 magnum opus on the topic, which described 100 cases and coined the term. Pondering neural mechanisms for the Parks syndrome, the first two clinical features listed in Table 8-1 are not difficult to explain. Receptive fields in V1, representing the fovea, are tiny and have narrow tolerances.1,39 Any defocusing or other decorrelation of one eye’s inputs produces a conflict in neighboring foveal V1 columns and promotes suppression of ODCs corresponding to the weaker eye. The fovea subtends approximately 5° of the retinotopic map of V1; thus a suppression scotoma of <5° (feature 1 of the syndrome) makes sense. Regional suppression of metabolic activity (an anatomic “suppression scotoma”) has been documented in V1 of strabismic monkeys.21,22 Feature 2, subnormal stereopsis, can be explained the same way. Stereoacuity decreases exponentially from the fovea to more eccentric positions along the retinotopic map of the visual field.40 If foveal ODCs are suppressed and parafoveal ODCs are left to mediate stereopsis, stereopsis is degraded but not obliterated. But it is features 3 and 4 of Parks’ list that are most intriguing. If binocular development is perturbed so that right- and left-eye foveal ODCs (receptive fields) do not enjoy correlated activity, why should the fall-back position of the visual cortex be set so predictably approximately 2° to 4° (approximately 4 to 8 PD) of microesotropia (Fig. 8.4A)? And if the heterotropia exceeds that range, why is fusional vergence typically absent? Studies of fusional anatomy and neurophysiology in primates have revealed candidate mechanisms.
Only gold members can continue reading. Log In or Register to continue
Jul 11, 2016 | Posted by drzezo in OPHTHALMOLOGY | Comments Off on Binocular Vision Adaptations and Maldevelopments in Monofixation Syndrome, Microstrabismus, and Macrostrabismus