Recurrent oral ulcers (at least three times per year) plus two of the following:
1. Recurrent genital ulcers (aphthous or scarring ulcers)
2. Ocular inflammation (anterior uveitis/posterior uveitis, cells in the vitreous on slit-lamp examination, or retinal vasculitis)
3. Skin lesions (erythema nodosum, pseudofolliculitis, papulopustular lesions, or acneiform nodules in postadolescent patients not on corticosteroid treatment)
4. Positive pathergy skin test (read by the physician at 24–48 h after a prick to the forearm by a sterile needle)
Etiopathogenesis
The etiopathogenesis of the disease still remains unknown; however, it is generally believed that it represents an enhanced or dysregulated immune response triggered by environmental factors in immunogenetically susceptible individuals [27]. Genetic, immunological, and environmental factors have been studied intensively. The most significant genetic association of Behçet disease is with the major histocompatibility antigen HLA-B51 [28]. Recently, genome-wide association studies identified single nucleotide polymorphism within the gene-encoding interleukin-10 (IL-10) and disease-associated nonsynonymous variants in the toll-like receptor 4 (TLR4) gene in patients with Behçet disease [29, 30]. Abnormal cellular immune responses including that of pathergy skin prick test induced sterile inflammation mediated by neutrophils, T-cell-mediated immune responses and abnormal response to bacterial antigens, increased Th 1 cytokine production, defects in the complement system, and upregulation of endothelial cell surface molecules have all been described [1, 19, 22]. Since Behçet disease manifests as an obliterative vasculitis, hemodynamics and coagulation factors have also been studied [19]. Environmental factors including infectious agents seem to play an important role in the pathogenesis. Individuals who have emigrated from endemic countries to historically non-Behçet regions of the world, such as the Turks living in Germany or the Japanese living in the United States, have a reduced risk [1, 19]. Furthermore, recent studies from Japan, where the disease is common, have shown a decreased incidence and severity of the disease despite stability in the genetic configuration of the population suggesting the role of environmental factors on the development of the disease [19, 31–37].
Ocular Involvement in Behçet Disease
Ocular involvement in Behçet disease is characterized by occurrence of relapsing remitting uveitis attacks of explosive nature and with spontaneous remissions. Behçet uveitis occurs in more than 50 % of the patients with Behçet disease in hospital cohorts, with a higher rate in male patients ranging between 70 and 90 % [1]. Behçet uveitis is the major cause of noninfectious uveitis in Turkey and accounts for one-third of uveitis cases seen at referral centers [38]. In a minority of patients, ocular involvement may present with conjunctival ulcers, episcleritis, scleritis, keratitis, isolated optic neuritis, and extraocular nerve palsies from neurologic involvement of Behçet disease [1, 39].
Behçet Uveitis
Behçet uveitis may involve the anterior or posterior segment of the eye, or more commonly both [1, 40]. The classification of uveitis as anterior or posterior is important both therapeutically and prognostically as lesions affecting the posterior segment are of persistent nature and are correlated with significant visual loss [41]. The most common presentation in Behçet uveitis is in the form of bilateral nongranulomatous panuveitis with retinal vasculitis [1, 40]. The disease may start as unilateral and/or anterior uveitis and become bilateral with posterior segment involvement [1].
Anterior Segment Inflammation
Anterior uveitis is typically nongranulomatous in nature and can occur as an isolated finding in about 10 % of patients [40]. Isolated anterior uveitis is a more common presentation in female patients [40, 42, 43]. The eye can appear white despite profound anterior chamber inflammatory reaction with hypopyon formation (Fig. 3.1). However, ciliary injection and conjunctival hyperemia may also be noted. The inflammatory reaction observed in the anterior chamber is characterized by absence of fibrinous exudation and presence of inflammatory cells that circulate freely. Because fibrinous exudation is uncommon, cells can form a smooth-layered hypopyon that shifts with gravity. The hypopyon in Behçet uveitis has a transient nature, and its presence strongly implies severe posterior segment involvement in the affected eye. The Systemic Immunosuppressive Therapy for Eye Diseases (SITE) Cohort Study retrospectively analyzed the prevalence and incidence of hypopyon in nearly 5000 patients with uveitis and found a fivefold increased risk of hypopyon in Behçet disease in a non-endemic population [44]. Although hypopyon itself is a nonspecific finding, it remains as the hallmark of the disease as part of the historical definition of Behçet uveitis as “iridocyclitis with hypopyon” [2]. Hypopyon formation has been reported to occur in 5–30 % of eyes [40, 42, 43, 45–47]; however, its true incidence may be higher than the reported figures as it may be missed if the patient does not present at the onset of a uveitis attack.
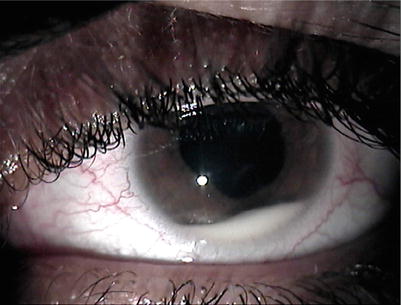
Fig. 3.1
Hypopyon in a patient with Behçet panuveitis. Please note that the eye is white without conjunctival and ciliary injection, representing the so-called cold hypopyon
Assessment of the anterior chamber flare by laser flare photometer in patients with Behçet uveitis appears to be a useful ancillary test as readings show a significant correlation with the anterior chamber cell score, vitreous haze score, and the presence of inflammatory retinal lesions and active retinal vasculitis during an acute inflammatory episode [48]. The anterior chamber flare score also shows correlation with the fluorescein angiographic leakage in eyes with clinical remission, and a flare reading more than 6 photons/ms is associated with an increased risk of a recurrent uveitis attack [48].
Posterior Segment Inflammation
Presence of diffuse vitritis, retinal perivasculitis, superficial and/or deep retinal infiltrates that are usually accompanied by optic disk inflammation and/or cystoid macular edema is characteristic features of posterior segment inflammation in Behçet uveitis. Spontaneous resolution of acute inflammatory signs in the posterior segment is a uniform feature and is a clue in differentiating Behçet uveitis from other forms of severe noninfectious and infectious posterior uveitides.
Diffuse cellular infiltration of the vitreous is a constant feature [40]. Vitreous haze may also be present depending on the severity of vitreous infiltration and is an indicator of active inflammation and is most severe at the onset of the uveitis attack. The vitreous inflammation may be so profound that it may lead to the loss of the fundus reflex [40]. During the resolution of diffuse vitritis, the cellular vitreous infiltration tends to precipitate in the inferior periphery of the retina appearing as inflammatory precipitates aligned in a stringlike configuration (Fig. 3.2) [40]. This finding is pathognomonic for Behçet uveitis and should not be confused with snowball opacities seen in intermediate uveitis. Inferior peripheral vitreous inflammatory precipitates appear several days after the onset of a Behçet uveitis attack and resolve spontaneously without leaving any sequelae.
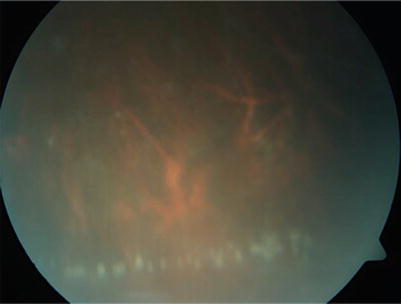
Fig. 3.2
Color fundus photograph of the inferior peripheral retina in a male Behçet uveitis patient who presented 3 days after the onset of uveitis attack. The view is blurry because of diffuse vitritis. Precipitated vitreous cells led to the appearance of inflammatory precipitates in the inferior periphery of the retina with a stringlike alignment
Retinal perivasculitis occurs in 80–100 % of patients and is characterized by segmental or diffuse perivenous fluffy white haziness surrounding the blood vessel [40, 42, 43, 45–47]. Retinal veins are more commonly affected than the arteries; however, retinal arteriolitis may accompany retinal periphlebitis, and retinal capillaritis may be evident on fluorescein angiography as diffuse capillary leakage in a fernlike pattern [49]. Periphlebitis may be difficult to visualize at the beginning of an acute inflammatory episode due to severe accompanying vitritis, but is easily detected as diffuse gliotic sheathing following the resolution of the perivascular inflammation [40]. Very severe breakdown of the blood-retina barrier may lead to a fundus appearance similar to frosted-branch angiitis with hemorrhages along the inflamed retinal vascular tree.
Occlusion of the retinal veins may occur at any location from the central retinal vein to the tiny small branches and is of recurrent nature. Retinal hemorrhages are frequently evident along the affected vein distribution (Fig. 3.3) [40]. After resolution of the periphlebitis, fluorescein angiography may show extensive retinal capillary nonperfusion, which may lead to neovascularization of the disk (Fig. 3.4) or elsewhere (Fig. 3.5) [50]. Although occlusive periphlebitis in Behçet uveitis is typically associated with diffuse vitritis, thrombotic retinal vascular occlusion may also develop in the absence of significant ocular inflammation. The latter finding has been attributed to elevated prothrombotic factors, prothrombin gene G20210A mutations, and elevated serum homocysteine levels in patients with Behçet disease [51].
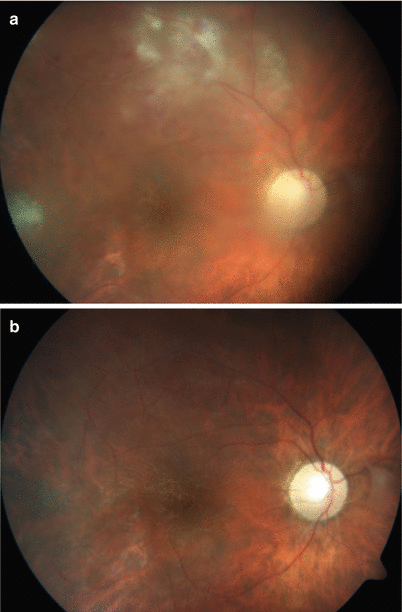
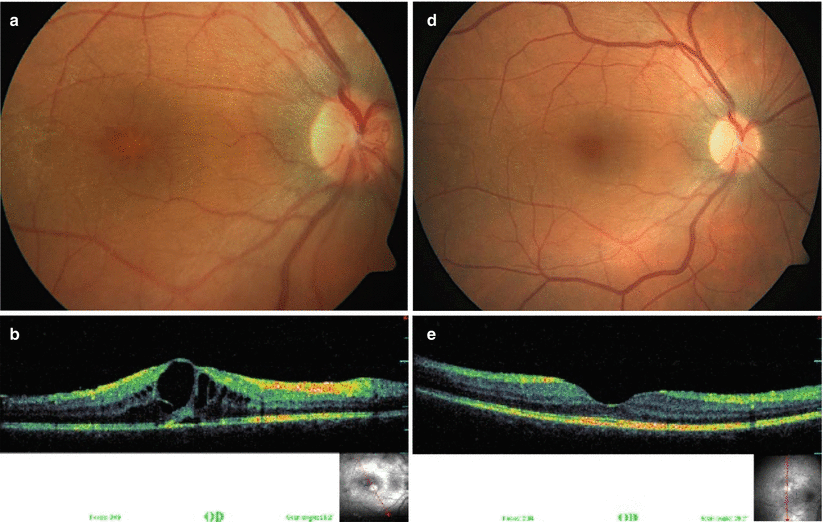
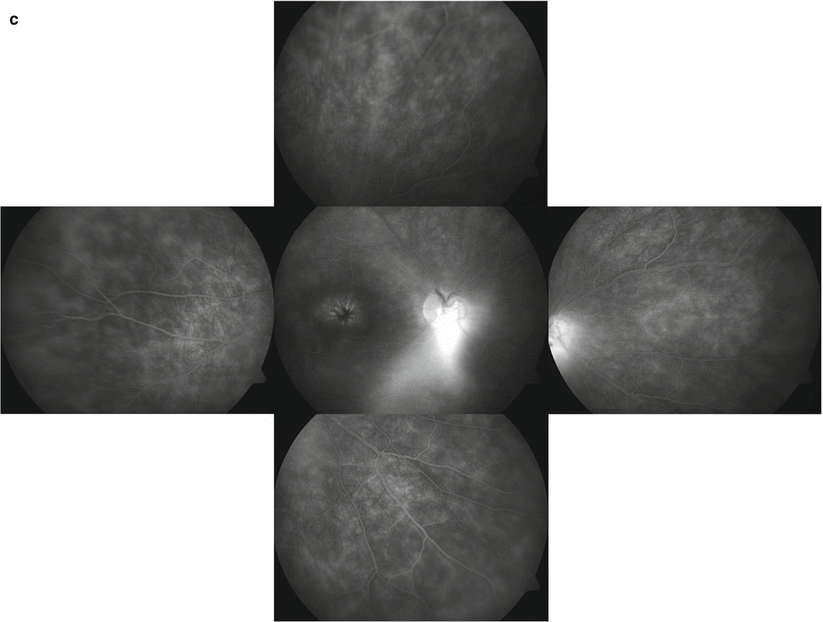
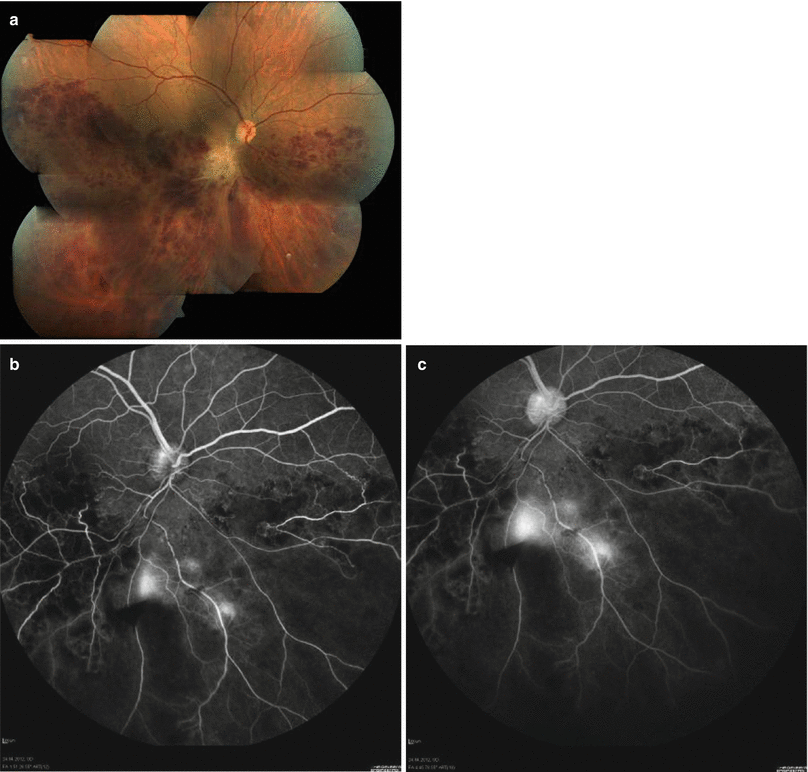
Fig. 3.3
Color fundus photograph shows retinal occlusive periphlebitis along the superior temporal vascular arcade in a male patient with Behçet panuveitis. Retinal hemorrhages are evident along the affected retinal vein. A retinal infiltrate is evident temporal to the macula. View of the posterior segment is blurry because of diffuse vitritis (a). Color fundus photograph of the same patient 3 weeks after increasing the dose of interferon alpha-2a to 6 million international units shows gliotic sheathing along the affected vein with resolution of diffuse vitritis, periphlebitis, retinal hemorrhages, and retinal infiltrate. Empty ghost retinal vessels are apparent at the disk margin, the optic disk shows pallor, the macula appears atrophic and degenerated, and total posterior vitreous detachment is evident (b)
Fig. 3.4
Color fundus photograph of a female Behçet uveitis patient shows optic disk neovascularization and cystoid macular edema (a). Spectral-domain optical coherence tomography (SD OCT) confirms the presence of cystoid macular edema (b). Late-phase fluorescein angiogram of the posterior pole shows profuse fluorescein leakage from the optic disk due to neovascularization and a petaloid fluorescein pattern associated with cystoid macular edema; diffuse capillary leakage without retinal ischemia is evident in the peripheral sweeps (c). Color fundus photograph (d) and SD OCT (e) obtained 8 months after institution of interferon alpha therapy show complete resolution of the disk neovascularization and the cystoid macular edema
Fig. 3.5
Color fundus photograph of a male patient with Behçet uveitis shows severe occlusive retinal periphlebitis with retinal hemorrhages along the branches of the inferior hemispheric retinal vein (a). Fluorescein angiogram obtained 11 weeks after the presentation shows fluorescein blockage due to retinal hemorrhages and areas of capillary nonperfusion along with three spots of fluorescein leakage in the venous phase of the angiogram (b). An increase in fluorescein leakage at the late phase (c) confirms the presence of retinal neovascularization secondary to retinal ischemia
Subclinical chronic retinal vasculitis may be demonstrated by fluorescein angiography, suggesting persistence of retinal vascular inflammation during apparently quiescent periods between attacks [49, 52]. Staining of the optic disk and retinal capillary leakage are the most common angiographic findings indicating ongoing subclinical inflammation in the posterior segment. Persistence of vitreous haze, optic disk hyperemia, and cystoid macular edema are the clinical signs of inadequately controlled intraocular inflammation (Fig. 3.6). Optic disk neovascularization may also develop as a result of persistent subclinical inflammation, and fluorescein angiography shows diffuse retinal vascular and capillary leakage in the absence of retinal ischemia. Besides clinical evaluation, therapeutic efficacy should be monitored by fluorescein angiography as absence of retinal vascular and capillary leakage is considered as a sign of complete remission.
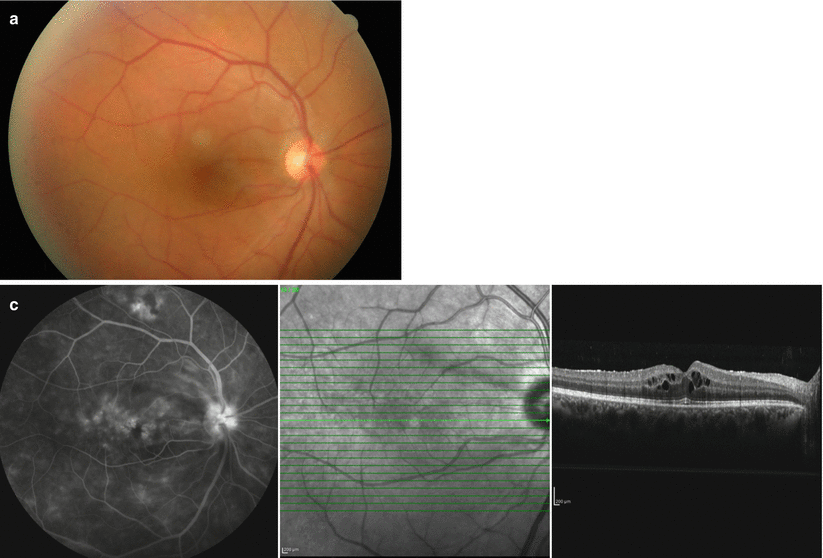
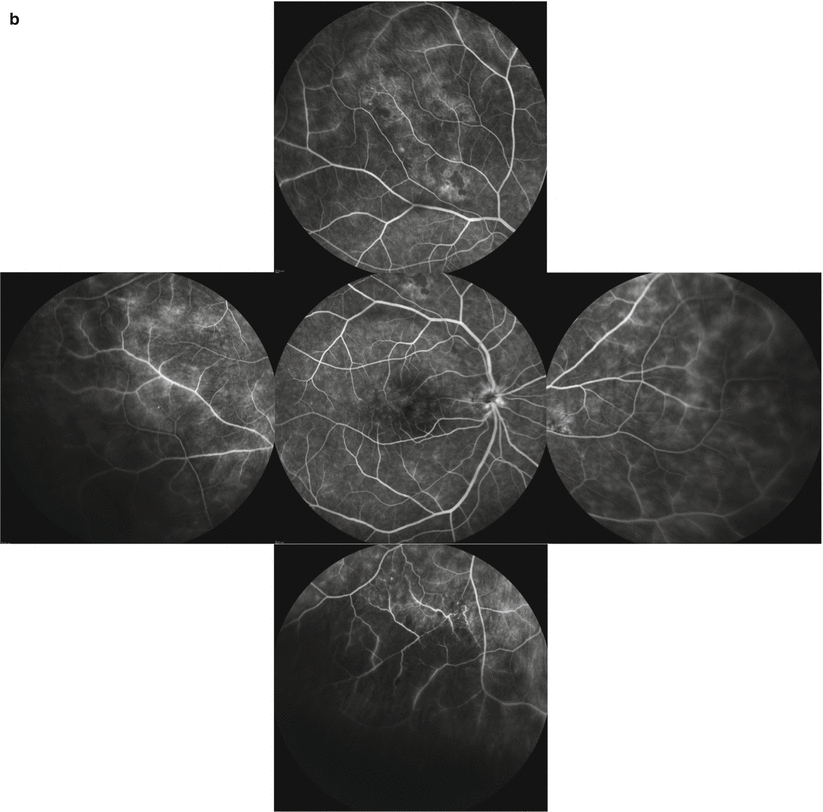
Fig. 3.6
Color fundus photograph of a male patient with Behçet uveitis shows no apparent signs of inflammation (a). Venous-phase fluorescein angiography of the posterior pole shows faint fluorescein leakage from the posterior pole and the optic disk; peripheral sweeps show areas of vascular and diffuse capillary leakage along with areas of capillary nonperfusion (b). Late-phase angiogram shows diffuse leakage from the posterior pole with a petaloid pattern at the fovea suggesting cystoid macular edema and optic disk staining. Spectral-domain OCT confirms the presence of cystoid macular edema and a very small serous retinal detachment (c)
Transient superficial retinal infiltrates occur during activation of uveitis and can involve any location and number and resolve within a few days without leaving any visible scar formation (Figs. 3.7 and 3.8). Deep retinal infiltrates may take longer to resolve and may leave scars. An unrecognized feature is the occurrence of retinal nerve fiber layer defects following resolution of retinal infiltrates at the posterior pole. They appear as wedge-shaped retinal nerve fiber defects in the peripapillary area (Fig. 3.9). Corollary thinning detected by optical coherence tomography and corresponding visual field loss occur in Behçet uveitis patients in the absence of central nervous system involvement or prior optic neuritis [53]. The neuroretinal rim is well preserved in these cases in contrast to the loss of neuroretinal rim associated with retinal nerve fiber atrophy in glaucoma. We hypothesize that these defects occur as sequelae of retinal infiltrates leading to an acute focal damage to the ganglion cell and the retinal nerve fiber layer.
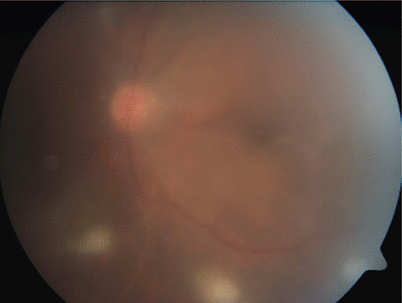
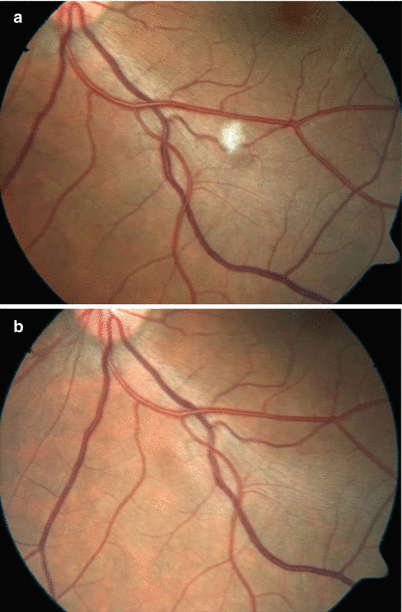
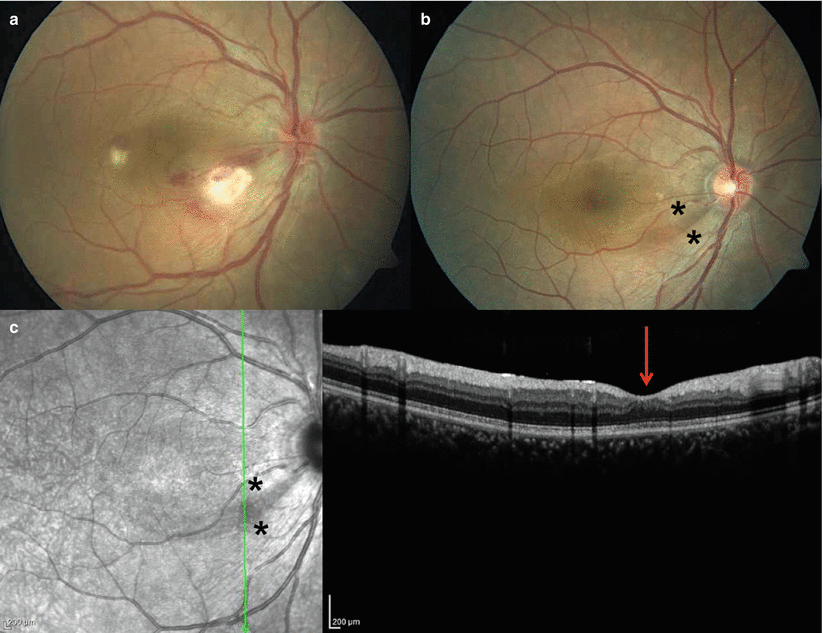
Fig. 3.7
Color fundus photograph of a male patient with Behçet panuveitis shows superficial retinal infiltrates at the posterior pole and the inferior retinal periphery; the view is blurry because of diffuse vitritis
Fig. 3.8
Color fundus photograph of a male patient with Behçet uveitis shows a superficial retinal infiltrate along the inferotemporal retinal vein (a) and its resolution within a few days without leaving any visible scar formation (b)
Fig. 3.9
Color fundus photograph of a male patient with Behçet uveitis shows a superficial retinal infiltrate on the inferotemporal vascular arcade with retinal hemorrhages, a second superficial retinal infiltrate is evident temporal to the fovea, and the optic disk is hyperemic (a). Color fundus photograph obtained during quiescence 33 months after the uveitis attack shows a wedge-shaped peripapillary nerve fiber layer bundle defect corresponding to the previous location of the retinal infiltrate and is demarcated by the asterisks (b). The green line on the scanning laser ophthalmoscope image shows the location of scan line through the nerve fiber bundle defect demarcated by the asterisks. Spectral-domain optical coherence B-scan reveals deficiency in the nerve fiber layer leading to an indentation (red arrow) in the inner retina as well as disfigurement of the inner retinal layers at the same location (c)
Complications
Recurrent inflammatory episodes lead to ocular complications of which macular edema and optic atrophy are associated with irreversible and profound vision loss [20]. The most common complications of Behçet uveitis are cataract, persistent posterior synechiae, macular edema, optic atrophy, epiretinal membrane formation, and glaucoma [40]. Disk and/or retinal neovascularization, macular hole, retinal detachment, and phthisis bulbi are rare complications [40]. Optical coherence tomography is a useful ancillary method in diagnosing and assessing the response to treatment of macular complications of Behçet uveitis such as macular edema, epiretinal membrane, macular hole, and foveal atrophy [53]. The end-stage fundus is characterized by optic atrophy, ghost retinal vessels, diffuse atrophy, and gliosis of the retina with variable pigmentation and macular scarring (Fig. 3.10). The vitreous appears clear at the end stage.
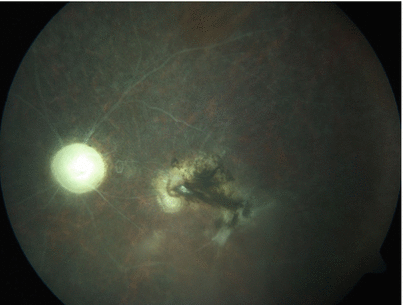
Fig. 3.10
Color fundus photograph shows end-stage fundus in a male patient with Behçet uveitis. Optic atrophy, retinal ghost vessels, diffuse atrophy and gliosis of the retina with variable pigmentation, and macular scarring are apparent, and the vitreous appears clear
Treatment of Behçet Uveitis
Patients with Behçet uveitis are no more treated with long-term high-dose systemic corticosteroids, which inevitably lead to severe unacceptable systemic side effects. Such therapy is also associated with a rebound effect during dose tapering and is not capable of preventing recurrences of uveitis attacks. Therefore, today, first-line therapy of Behçet uveitis involves the use of immunomodulatory agents in an effort to attain a steroid-sparing effect and to prevent recurrences of uveitis attacks in an effective manner. Overall, main treatment goals of Behçet uveitis include (1) rapid control of acute inflammatory episodes, (2) prevention of recurrences, and (3) achievement of durable remission after discontinuation of treatment.
Acute inflammatory episodes are still treated with corticosteroids, despite the lack of randomized controlled trials that show their efficacy on the resolution of active intraocular inflammation. High-dose systemic corticosteroids are used to control severe posterior or panuveitis attacks that pose a threat to vision. Intravenous pulse methylprednisolone (1 g/day, a single dose or for 3 consecutive days) is usually administered to obtain a rapid anti-inflammatory effect [54, 55]. Oral prednisone 1 mg/kg/day or its equivalent is then given and slowly tapered to a maintenance dose of 7.5 mg/day or less after complete resolution of active inflammation. An adjunct periocular or intravitreal corticosteroid injection may be used for the treatment of unilateral severe panuveitis attacks and/or cystoid macular edema especially when high-dose systemic corticosteroid treatment is ineffective or contraindicated [56, 57]. Recently, in a pilot study, a single infusion of intravitreal infliximab has also been reported to be efficacious in controlling a sight-threatening attack of Behçet uveitis [58]. In retrospective and prospective clinical studies, a single infusion of infliximab at a dose of 5 mg/kg has been shown to be effective in rapidly controlling an acute posterior or panuveitis attack due to Behçet disease [59–61]. In a nonrandomized clinical study, a single infusion of infliximab has also been shown to be faster in controlling an acute inflammatory episode than intravenous or intravitreal corticosteroids [62]. For the treatment of isolated anterior uveitis attacks, frequent application of potent topical corticosteroids with a tapering schedule over 6–8 weeks is usually sufficient. Topical mydriatic and cycloplegic agents should simultaneously be added twice or thrice daily during the acute phase for a period of 2–3 weeks.
In patients with Behçet uveitis, posterior segment involvement is an absolute indication for the use of immunomodulatory therapy. An expert panel has advocated systemic immunomodulation as soon as the patient has been identified to have posterior segment inflammation due to Behçet disease [63]. Furthermore, based on systematic review of the literature, the European League Against Rheumatism proposed that Behçet uveitis patients with posterior segment involvement should be started on azathioprine and systemic corticosteroids, and patients with severe eye disease (defined as those who have more than two lines of drop in visual acuity or those with retinal vasculitis or macular involvement) should have either cyclosporine A or infliximab added to their regimen or should receive interferon alpha instead [64].
Azathioprine and cyclosporine A have long been used in the long-term management of Behçet uveitis. Their efficacy in the treatment of Behçet uveitis has been proven in randomized clinical trials [65–67]. Combination therapy with azathioprine and cyclosporine A appears to be more effective than monotherapy with either agent alone. Because of potential side effects, there is need for regular follow-up of complete blood count (CBC) and liver function test with azathioprine and CBC and kidney function tests and blood pressure monitoring with cyclosporine A. Cyclosporine A should not be used in patients with concomitant neurologic involvement [68]. Mycophenolate mofetil or mycophenolate sodium, which is better tolerated due to less gastrointestinal side effects, has been shown to be effective as a steroid-sparing agent in uncontrolled studies that included also patients with Behçet uveitis [69, 70]. Tacrolimus, another calcineurin inhibitor as cyclosporine A, is associated with less severe side effects; however, data is lacking, as it has not been widely studied for the treatment of Behçet uveitis [71, 72]. Colchicine is an anti-inflammatory agent that is effective for controlling mucocutaneous manifestations of Behçet disease; however, its efficacy has not been proven for ocular involvement [22, 73].
The use of alkylating agents is now limited in the management of Behçet uveitis because of their potential side effects including an increased risk of malignancy and because of the introduction of biologic agents into our armamentarium. One comparative study suggested that cyclosporine was superior to pulsed intravenous cyclophosphamide especially during the initial 6 months of therapy [67]. Although open studies with chlorambucil have shown some efficacy, suggesting even a durable remission after its discontinuation, it had been used only as a last resort until the biologic agents became available [74, 75].
In the 2000s, biologic agents have become widely used for the treatment of Behçet uveitis. These agents specifically target the components of the immune system responsible for dysfunction. Interferon alpha-2a has been shown to be effective with a response rate ranging between 85 and 98 % in numerous studies [76–86]. The initial report by Kötter and associates involves the use of interferon alpha-2a starting at a dose of 6 million international units (IU) per day for 7 days and adjusting the dose based on the response over the initial 28 days [76]. There is no consensus about the optimum dose and duration of interferon alpha-2a therapy in Behçet uveitis. Low-dose and dose-escalating protocols have been proposed with the rationale of avoiding side effects of interferon alpha-2a therapy, some of which are dose dependent [82, 84]. Side effects of interferon alpha-2a therapy include flu-like symptoms experienced almost by all patients at the beginning of therapy and with high-dose maintenance injections, leukopenia, thyroiditis, occurrence of autoantibodies (usually without clinical autoimmune disease), alopecia, elevated liver enzymes, weight loss, and depression [76, 77]. The main advantage of interferon alpha therapy over other agents seems to be its ability to induce durable remission after treatment discontinuation [86].
Infliximab is a human-murine chimeric monoclonal antibody against tumor necrosis factor (TNF) alpha. A number of open, prospective self-controlled trials have shown efficacy of repeat infliximab infusions in decreasing the number of uveitis attacks, reducing the dose of corticosteroids and in preserving or improving the visual acuity in refractory cases of Behçet uveitis [60, 87–94]. One retrospective comparative study revealed fewer attacks, better visual acuity, and fewer ocular and systemic complications among those patients treated with infliximab than those receiving conventional immunomodulatory therapy [95]. A more recent comparison between infliximab and cyclosporine in the treatment of Behçet uveitis looked specifically at the initial 6 months of treatment with each, and the 6-month period before and after treatment was compared within and between the study groups. Both therapies significantly decreased the number of acute attacks, and patients treated with infliximab experienced significantly fewer attacks during treatment than patients treated with cyclosporine, but no significant difference in visual acuity improvement was evident between the two groups [96]. Repeat infusions are associated with the occurrence of neutralizing antibodies that lead to decreased serum concentration of infliximab eventually resulting in recurrence of uveitis and need for reduced interval between infusions [97]. It is recommended to combine infliximab with low-dose conventional immunomodulatory agents in an effort to preserve its efficacy. Discontinuation of infliximab therapy seems to be associated with a high relapse rate [87, 88]. Side effects are mostly mild with infusion reaction appearing as the most common one. Although rare, reactivation of pulmonary tuberculosis remains a major concern in countries where tuberculosis is endemic [87, 91]. Despite the fact that etanercept, a soluble TNF alpha receptor, has been reported to be ineffective in the treatment of endogenous uveitis and that it may even lead to new onset of uveitis in patients with other rheumatological diseases [98, 99], a beneficial but unsustained effect has been reported in one study of refractory Behçet uveitis patients [100]. There are only limited reports with limited number of patients on the efficacy of adalimumab, a humanized monoclonal anti-TNF antibody, in patients who are refractory to other immunomodulatory agents including infliximab [101–106]. Other biologic agents that have been reported to be effective in case reports or small pilot studies of Behçet uveitis include golimumab, a fully human anti-TNF monoclonal antibody with reduced immunogenicity and longer half-life; gevokizumab, an IL-1beta regulating monoclonal antibody; rituximab, a chimeric monoclonal antibody against B-cell antigen CD20; and tocilizumab, an anti-IL-6 receptor antibody [107–111]. In randomized controlled trials, daclizumab, an anti-IL-2R antibody, and secukinumab, an anti-IL-17A antibody, have not been found to be effective in the treatment of Behçet uveitis [112, 113].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


