Purpose
To assess the visual prognosis of patients with ocular Behçet disease and to determine factors predictive of visual loss and severe visual loss.
Design
Retrospective case series.
Methods
One hundred seventy-five eyes of 107 patients diagnosed with ocular Behçet disease were included. The main outcome measures were visual loss (best-corrected visual acuity, worse than 20/40) and severe visual loss (best-corrected visual acuity, 20/200 or worse).
Results
The mean duration of follow-up was 6.5 years. Presenting visual acuity was worse than 20/40 in 50% of eyes and 20/200 or worse in 21% of eyes; approximately one third of this was reversible with treatment. The most common cause of irreversible severe visual loss was ischemic maculopathy. At 10 years, there was a 39% risk of visual loss and a 24% risk of severe visual loss, the latter figure being reduced to 13% if patients with irreversible visual loss at presentation were excluded. After controlling for potentially confounding variables, male sex, unilateral disease, and left eye involvement all were statistically significant risk factors for severe visual loss at 5 and 10 years. Patients who were treated with biologic agents were less likely to have severe visual loss in either eye at both 5 and 10 years.
Conclusions
Many patients with ocular Behçet disease still have irreversible visual loss at presentation. However, the visual prognosis is otherwise improved, with a 10-year risk of severe visual loss of 13% in this cohort. The use of biologic agents is associated with a lower risk of severe visual loss at 5 and 10 years.
Behçet disease is a long-term, multisystem disorder characterized by relapsing inflammation. Ocular involvement occurs in approximately 70% of patients, typically in the form of a relapsing-remitting uveitis with a significant ischemic element. It has a high prevalence among people with ancestors located along the old Silk Road, but is an important cause of morbidity throughout the world. Behçet disease typically affects young adults and is said to be more common in men, although the sex ratio varies depending on geographic location—the disease is more common in women in Western Europe and the United States, and a recent Spanish series even showed a slight preponderance of women. Nevertheless, there is general agreement that men have more severe disease than women, including the risk of developing significant ocular inflammation. Male patients tend to have a younger age of onset, which is associated with a higher prevalence of ocular disease and a worse potential visual acuity at presentation, although age and sex both have been shown to be independent risk factors for poor visual outcome. Behçet disease also tends to be more aggressive in Eastern than Western populations, probably as a result of genetic heterogeneity.
Ocular inflammation often is present at the outset of Behçet disease and is the initial manifestation in approximately 20% of cases. If not present at disease onset, ocular involvement occurs most commonly within 2 to 4 years, eventually affecting more than 50% of patients, including 70% to 90% of men. Anterior uveitis is more common in women, and posterior uveitis is more common in men. The visual outcome of Behçet disease is generally poor, although it is improving. Initial work by Ben-Ezra and Cohen suggested that severe visual loss occurred in 74% of patients within 6 to 10 years. It also has been reported that 50% of patients become blind within 5 years if untreated. The prevalence of legal blindness in Behçet disease patients was 25% in the United States in 1980s, and legal blindness also has been reported to ensue within 4 years of onset in 50% to 90% of patients in Japan and Turkey, although the advent of newer therapies may be improving the prognosis. Indeed, survival analysis of a relatively recent large series suggested that the risk of visual loss increases progressively, reaching 25% of cases by 10 years, but then remains relatively constant. A report from the Systemic Immunosuppressive Therapy for Eye Diseases Cohort Study Group recently suggested an incidence rate of severe visual loss of 0.09 per eye-year, although the median follow-up in this study was only 1.05 years.
In this study, we aimed to assess the visual prognosis for patients with Behçet disease in an era in which new intraocular and systemic therapies have become more widely available, including the use of the so-called biologic agents directed against tumour necrosis factor α (TNF-α). We retrospectively analyzed the demographic and clinical features, visual outcomes, and treatment of patients with ocular Behçet disease seeking treatment at 2 tertiary referral clinics over the last 10 years. We then compared the prognosis of our patients with that of previously reported studies to see whether this has improved and whether we could identify factors still associated with poor visual outcome.
Methods
We performed a retrospective study of 107 patients with ocular involvement of Behçet disease who visited tertiary referral uveitis clinics at Moorfields Eye Hospital, United Kingdom, and Sydney Eye Hospital, Australia, between 2000 to 2010 and were followed up for at least 6 months. Only patients who met the 1990 classification criteria of the International Study Group for Behçet Disease were included in the study. We are currently involved in a randomized controlled trial of interferon-α in Behçet disease and did not include any of the data obtained within the controlled trial in this study, but did include patients up to the point at which they entered the trial.
Patients were managed with oral prednisolone and second-line immunosuppressive agents according to the guidelines for the use of immunosuppressive drugs in patients with ocular inflammatory disorders. Biologic agents were considered if conventional immunosuppressive agents failed and were administered in 28 of 107 patients. Biologic agents used included infliximab, etanercept, and adalimumab; there was no recorded use of interferon in the patients in this study. Local steroid delivery (including orbital floor steroid injection of 40 mg methylprednisolone acetate and intravitreal injection of 4 mg triamcinolone acetate) was considered for unilateral disease activation or in the presence of a contraindication to an increase in doses of systemic medication.
A complete ocular examination was performed at each visit, including best-corrected visual acuity on a Snellen chart, slit-lamp biomicroscopy, and tonometry. Indirect ophthalmoscopy was performed where indicated. For the purpose of the study, a data form was prepared that included demographic data such as sex, age at presentation, extraocular clinical manifestations, and immunosuppressive therapy. We do not routinely perform human leukocyte antigen (HLA) typing in suspected Behçet disease. Disease duration was defined as the time between the diagnosis of Behçet disease and the first presentation to the clinic with ocular involvement. Initial visual acuity was defined as the best-corrected visual acuity obtained at the initial clinic visit. In accordance with the standardization of uveitis nomenclature (SUN) criteria, patients were classified as having visual loss at a visual acuity of worse than 20/40 and severe visual loss at a visual acuity of 20/200 or worse. The time point at which visual loss occurred was defined as the time at which visual acuity decreased to worse than 20/40 and did not recover; severe visual loss was defined similarly, but with a visual acuity of 20/200 or worse. For visual loss to be classified as irreversible, patients had to have received at least 6 months of treatment without the occurrence of visual recovery.
For the analysis of data, a database was constructed using Microsoft Excel 2007 (Microsoft, Redmond, Washington, USA). Snellen acuities were converted into their logarithm of the minimal angle of resolution (logMAR) equivalent for analysis. The Mann–Whitney U test was used to compare means, and the Wilcoxon matched-pairs signed-rank test was used to compare visual acuities over time. The Fisher exact test was used to compare proportions, and the Kaplan-Meier method was used to estimate survival curves, with the log-rank test being used to perform comparisons between different survival curves. Variable frequency tables were tabulated both for patients and for affected eyes. By-person and by-eye analyses were performed to evaluate potential risk factors for visual loss, the former evaluating potential risk factors for visual loss in either eye. Risk factors controlled for in these analyses included: age, sex, race, duration of disease, multiple organ involvement (excluding aphthous ulcers), laterality of disease, presentation with ischemic complications, use of azathioprine, and use of biologic agents directed against tumour necrosis factor α. For these risk factor analyses, crude and adjusted odds ratios (ORs) were calculated using univariate and multivariate logistic regression, including 95% confidence intervals (CIs). For the multivariate logistic regression analyses, all statistically significant variables ( P < .05) from the univariate analyses were included as well as variables that were thought to be potential confounders. Analyses and graphs were performed using Microsoft Excel 2007, GraphPad Prism software version 5.01 (GraphPad Software, Inc., La Jolla, California, USA), and PASW Statistics software version 18.0.0 (IBM SPSS Inc., New York, New York, USA). P < .05 was accepted as being statistically significant.
Results
Baseline Patient Characteristics
A total of 107 patients (175 affected eyes) were included in the study. There were 57 male patients (53%) and 50 female patients (47%); the male-to-female ratio was 1.14:1. Sixteen patients were Afro-Caribbean (15%), 5 were Asian (5%), and the remainder were white (80%). The mean age at presentation to our clinics was 35.3 years (range, 17 to 63 years). The most common extraocular disease manifestation was recurrent oral ulcers (100%), followed by genital ulcers (51%) and arthritis (51%); central nervous system (19%), and gastrointestinal involvement (4%) were less common. The mean disease duration at the time of ocular presentation was 4.0 ± 0.63 years (mean ± standard error of the mean [SEM]; range, 0 to 31 years). The mean duration of follow-up in our study was 6.45 ± 0.34 years (mean ± SEM; range, 0.5 to 10 years), with 66 patients having follow-up of more than 5 years. Patients treated with anti–TNF-α agents had similar follow-up periods, with a mean duration of follow-up of 7.4 ± 0.46 years (mean ± SEM; range, 2.5 to 10 years).
Of the 107 patients, 68 had bilateral ocular disease, 27 had unilateral left eye disease, and 12 had unilateral right eye disease. Most patients who had bilateral disease had evidence of this at presentation—only 9 of the 68 patients with bilateral disease initially sought treatment with unilateral ocular involvement, and in these patients, second eye involvement developed at a median 6 months after first presentation (range, 0.6 to 2 years). Similar to the findings from a large series of Turkish patients reported in 2004, the proportion of patients with unilateral disease remained constant over the course of the study, at 37% of patients with up to 5 years of follow-up and 35% of patients with more than 5 years of follow-up ( P = 1.00). There was no evidence that azathioprine protected against second-eye involvement, as has been suggested previously: only 13 of 39 patients with unilateral disease who did not go on to develop second eye involvement were treated with azathioprine, compared with 4 of 9 patients in whom second eye involvement did develop ( P = .70).
Presenting Visual Acuity, Initial Response to Treatment, and Rates of Visual Loss
Ocular disease manifestations at presentation are included in Table 1 . Presenting visual acuity was less than 20/40 in 87 (50%) of 174 eyes, and 20/200 or less in 36 (21%) of 174 eyes. By 6 months, this had improved to 70 eyes (40%) and 23 eyes (7%), respectively, suggesting that approximately one third of the initial visual loss was reversible with treatment. When mean acuities were assessed over time, there was, therefore, a significant initial improvement over the first 12 months from 0.21 ± 0.03 logMAR to 0.19 ± 0.03 logMAR (mean ± SEM; P = .02), followed by a slow decline over time ( Figure 1 , Top). Of the 23 eyes that did not improve to better than 20/200 by 6 months, the most common cause of severe visual loss was ischemic maculopathy secondary to branch retinal vein occlusion (12 patients).
| Manifestation | No. (%) of 107 Patients | |
|---|---|---|
| At Presentation | With Permanent SVL | |
| Posterior/panuveitis | 79 (74) | 0 |
| Branch retinal vein occlusion | 30 (28) | 12 (11) |
| Anterior uveitis | 28 (26) | 0 |
| Ischemic retinal vasculitis | 23 (21) | 4 (4) |
| Optic nerve involvement | 7 (7) | 3 (3) |
| Central vein occlusion | 4 (4) | 3 (3) |
| Scleritis | 3 (3) | 0 |
| Central retinal artery occlusion | 1 (1) | 1 (1) |
| Retinal detachment | 1 (1) | 0 |
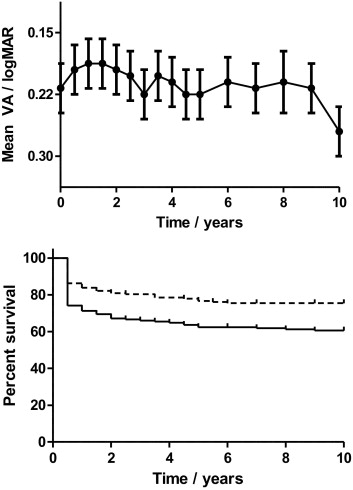
Kaplan-Meier survival analysis estimated the risk of visual loss to be 39% and the risk of severe visual loss to be 24% at 10 years ( Figure 1 , Bottom). This is similar to the 25% of patients with severe visual loss at 10 years reported in the series by Tugal-Tutkun and associates, but that analysis excluded patients with irreversible visual loss at presentation. Excluding these patients reduced the estimated risk of severe visual loss in our series to 13% at 10 years.
Male Versus Female Sex
There is general agreement that ocular Behçet disease is worse in men, and this also proved the case in our series. Male patients sought treatment a younger age than female patients (33.0 ± 1.3 years vs 37.4 ± 1.5 years; P = .05). Males also sought treatment with a lower mean visual acuity (0.33 ± 0.03 logMAR vs 0.17 ± 0.03 logMAR; P = .11) and with a shorter duration of systemic disease (2.99 ± 0.70 vs 5.16 ± 1.10 years; P = .09), although neither of these reached statistical significance. Males were not more likely to have bilateral disease ( P = .19), but were significantly more likely than females to have posterior uveitis rather than anterior uveitis alone (88% vs 66%; P < .01). Male patients also had a much worse prognosis in our series, Kaplan-Meier analysis estimating the risk of severe visual loss at 10 years to be 36% in men versus 14% in women (log-rank, 24.87; P < .01; Figure 2 , Top).
Laterality of Ocular Involvement
Whether patients had unilateral or bilateral ocular involvement influenced their chances of developing visual loss in an affected eye. Patients with unilateral disease did not have a significantly lower presenting visual acuity in the affected eye than those with bilateral disease (0.36 ± 0.05 logMAR vs 0.25 ± 0.03 logMAR; P = .07), but Kaplan-Meier survival analysis indicated that the risk of visual loss at 10 years was 56% for affected eyes in patients with unilateral disease versus 35% for eyes from patients with bilateral disease, and the risk of severe visual loss was 45% versus 19%, respectively (log-rank, 18.9; P < .001; Figure 2 , Middle). We considered that this could be the result of second-eye involvement being attenuated if second eye disease developed while already receiving treatment for first eye involvement, but 59 of 68 patients had bilateral disease at presentation, and removal of the data from the 9 patients in whom second eye involvement developed during the course of the study did not affect the statistical significance of the results. There was also no evidence that patients with unilateral disease were treated less aggressively than patients with bilateral disease, the mean number of immunosuppressive medications being 2.54 ± 0.24 for patients with unilateral disease versus 2.95 ± 0.18 for patients with bilateral disease (mean ± SEM; P = .45). Relapse rates also were similar in each group.
Interestingly, there was also a marked difference between the presenting visual acuity of affected right and left eyes, the former having a statistically significantly better acuity of 0.17 ± 0.03 logMAR as compared with 0.26 ± 0.03 logMAR ( P < .01); this difference was maintained over the 10-year follow-up period ( P < .01). Kaplan-Meier survival analysis gave a similar result, the risk of severe visual loss at 10 years being 20% for right eyes versus 29% for left eyes (log-rank, 9.66; P = .02; Figure 2 , Bottom).
Risk Factors for Visual Loss and Severe Visual Loss
We first looked for demographic and clinical characteristics associated with severe visual loss (visual acuity of 20/200 or less), and these are summarized in Table 2 . In the univariate analyses, only male sex was a statistically significant risk factor for the occurrence of severe visual loss in at least 1 eye at both time points. After controlling for potentially confounding variables, male sex, unilateral disease, and left eye involvement all were statistically significant risk factors for severe visual loss at 5 and 10 years; nonwhite race was a significant risk factor at 5 years, but not at 10 years. Patients who were treated with anti–TNF-α agents were statistically less likely to have severe visual loss in either eye at both 5 and 10 years. In contrast, use of azathioprine was not associated with a reduction in the risk of severe visual loss at 5 or 10 years. Analyses that evaluated potential risk factors for severe visual loss in any affected eye (a by-eye analysis) yielded similar results, although left eye involvement and nonwhite race were now no longer statistically significant ( Table 3 ).
| Risk Factor | Unadjusted 5-Year | Adjusted 5-Year | Unadjusted 10-Year | Adjusted 10-Year | ||||
|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P Value | OR (95% CI) | P Value | OR (95% CI) | P Value | OR (95% CI) | P Value | |
| Age | 0.98 (0.94 to 1.03) | .43 | 0.99 (0.95 to 1.04) | .68 | ||||
| Duration of disease | 1.02 (0.96 to 1.09) | .46 | 1.04 (0.97 to 1.11) | .30 | ||||
| Male sex | 3.61 (1.43 to 9.14) | < .01 a | 4.99 (1.73 to 14.4) | < .01 a | 2.90 (1.08 to 7.76) | .03 b | 5.42 (1.61 to 18.2) | < .01 a |
| Unilateral disease | 1.45 (0.60 to 3.52) | .41 | 3.31 (1.04 to 10.5) | .04 b | 1.67 (0.63 to 4.43) | .30 | 3.88 (1.04 to 14.49) | .04 b |
| Left eye involved | 3.30 (0.67 to 16.3) | .14 | 9.01 (1.37 to 60.5) | .02 b | 3.96 (0.74 to 21.1) | .11 | 15.8 (1.92 to 129.3) | .01 b |
| Nonwhite race | 2.69 (0.91 to 7.92) | .07 | 4.07 (1.11 to 15.0) | .04 b | 2.00 (0.61 to 6.58) | .25 | 3.66 (0.82 to 16.3) | .09 |
| Ischemic presentation | 1.08 (0.45 to 2.59) | .87 | 1.58 (0.61 to 4.08) | .35 | ||||
| Multiple organ involvement | 0.67 (0.27 to 1.69) | .40 | 0.83 (0.31 to 2.25) | .72 | ||||
| Treatment with azathioprine | 1.28 (0.54 to 3.06) | .59 | 1.67 (0.63 to 4.43) | .30 | ||||
| Treatment with biologic agents | 0.43 (0.17 to 1.12) | .09 | 0.28 (0.09 to 0.88) | .03 b | 0.31 (0.10 to 0.98) | .05 b | 0.18 (0.05 to 0.72) | < .01 a |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


