Basics of Inheritance
Arlene V. Drack
Most physicians think of inherited eye diseases as “zebras”—rare, one of a kind disorders seldom encountered in a typical practice. In fact, most patients seeking ophthalmologic care in industrialized nations have an inherited eye disorder. A typical patient’s axial length, corneal curvature, lens shape, and age at loss of lens clarity, as well as their predisposition to develop lattice degeneration, glaucoma, or age-related macular degeneration, are largely directed by their genes, as surely as is the more dramatic and devastating photoreceptor cell death in retinitis pigmentosa. From refractive errors and strabismus to cataracts and retinal detachments, ophthalmologists are in the business of treating genetic eye disease, often without realizing how current molecular genetic knowledge might aid in their diagnosis and treatment, or how further evaluation of certain patients and their families might contribute to our understanding of these disorders.
For centuries, ophthalmologic diagnosis has been a clinical and descriptive science. “Lumpers” argue with “splitters” over the significance of flecks and patterns in the retina and small differences in various parameters between groups of patients with similar disorders. The basic assumption has been that an observant, astute clinician can make accurate diagnoses on clinical grounds alone.
With the advent of molecular medicine, however, a paradigm shift is in order. For the first time in history we can offer some patients a blood test for gene mutation analysis that will tell us without a doubt what the diagnosis is. In the case of autosomal-dominant retinitis pigmentosa (ADRP), approximately 33% of patients can now receive a molecular diagnosis of either a rhodopsin or peripherin gene mutation.
For some mutations, a prognosis for progression can be given. Family members can have a blood test to determine whether they carry the gene. The same is possible for many patients with retinoblastoma, allowing children at risk to be identified early and treated at a stage when their vision and eye, as well as their life, often can be salvaged. Perhaps most illustrative of the power of this technique is the story of “butterfly” or “pattern” macular dystrophy. This entity was first described by Deutmann and associates in 19701 and has since become a “waste-basket” term used to describe different entities. In 1993, several groups of investigators2,3,4 discovered that novel mutations in the peripherin or RDS gene, a gene already known to cause ADRP, caused butterfly macular dystrophy in several large pedigrees. Once all family members had been genotyped, it was apparent that there was great phenotypic variation among persons with the same gene even in the same family. Some older members appeared to have age-related macular degeneration or Best’s disease-type lesions, whereas their siblings had typical butterfly lesions. One man with 20/200 vision had a lesion very similar to that of his sister, who had 20/25 vision and was almost the same age.
Studies such as these made it clear that the same genetic mutation can cause a wide range of clinical pictures even in the same family, whereas mutations of different genes, or different mutations of the same gene, can cause identical clinical pictures as in ADRP, in which many different mutations in the rhodopsin gene give the same clinical picture (i.e., bone spicules, disc pallor and flat ERG). Thus, we are at the dawn of a new era in which clinical appearance and tentative clinical diagnosis will indicate which genes should be tested, but the final diagnosis will be made on a molecular basis. A more refined and accurate nomenclature will be developed, and clinical trials will have the advantage of certainty that all patients in the study group actually have the same disorder. To offer their patients state-of-the-art care and current information, practitioners must acquaint themselves with this new form of diagnosis. In doing so, all physicians become part of the knowledge-gathering phase of this exciting and hopeful new era in ophthalmology.
OVERVIEW OF GENETIC TRANSMISSION
Most organisms, including humans, store and transmit genetic information via deoxyribonucleic acid (DNA). DNA is a long, linear polymer composed of many functional units called genes. The DNA of each gene makes a specific ribonucleic acid (RNA) by a process called transcription, which then directs production of a protein by a process called translation: DNA makes RNA makes protein. The protein may serve to maintain the cell in which it is made, or may be transported out of the cell to build or sustain body structures. The genes, together with certain proteins, are organized into rod shaped structures called chromosomes. When a cell divides, the DNA becomes more compact and the chromosomes can be seen under a light microscope. Normal human cells contain 46 chromosomes. This “compacting” during cell division is not for gene function, but rather to assist in the orderly and equal distribution of DNA between daughter cells.
DNA
The structure of DNA is a double helix with four bases: adenine, guanine, cytosine, and thymine. These are paired by hydrogen bonds between the two helices like rungs in a ladder (Fig. 1). Each base is associated with a sugar and phosphate forming a nucleotide. The nucleotides are numbered in order, giving the DNA sequence. Nucleotides in the linear polymer chains that make up the two helices are connected by phosphodiester bonds. The sequence of bases (in nucleotide form), in groups of three called codons, makes up the genetic code. Each triplet codes for a specific amino acid. Since 64 different triplets are possible from the four bases, and there are only 20 amino acids, more than one triplet can code for a given amino acid.5 For example, the codon AAA will always and only produce the amino acid phenylalanine in the protein, but other codons may also produce phenylalanine in the protein. Amino acids are ultimately arranged into proteins, the building blocks of living structures.
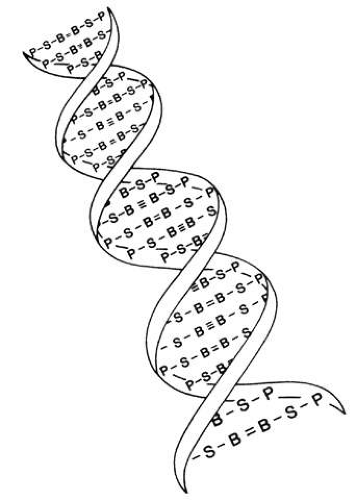 Fig. 1. Drawing of the DNA double helix, which is held together by hydrogen bonds between the bases (B). The other two units in the DNA molecule are deoxyribose sugar (S) and phosphate (P). |
In 1911, Johannsen coined the term “gene” as the basic unit of hereditary characteristics.6 In 1901, the monk Gregor Mendel described the dominant and recessive patterns by which some traits are inherited, leading to the term Mendelian inheritance.7 Garrod developed the concept of human biochemical disease (i.e., inborn errors of metabolism) and its inheritance in a recessive Mendelian fashion in 1908 and8,9
Genes are arranged into coding sequences, called exons, and intervening sequences that do not code, called introns. In addition, there may be separate promoter regions. Only 3% to 5% of human DNA codes for proteins; the rest has no known function.10 Although variation (mutation) in exons is likely to cause disease and is therefore rare, variation in introns does not cause a problem, yet is inherited in a Mendelian fashion. Since these variations are benign, they are also plentiful and vary from family to family. These variations are therefore used as genetic markers (e.g., in linkage analysis).
DNA sequences are written in a 5′ to 3′ direction (Fig. 2), and the sequence given is for the sense strand. The other strand is complementary and is called the template strand or antisense strand and runs in the opposite direction.10 DNA is transcribed in the following manner:
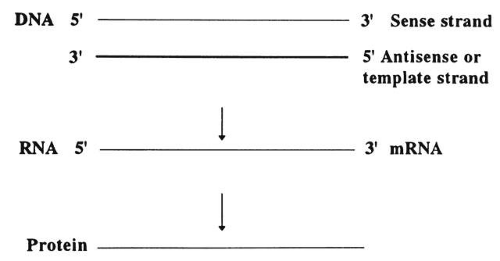 Fig. 2. DNA sequences for the sense strand (5′ and 3′ directions). In the double helix, the two strands run in opposite directions. Synthesis proceeds from 5′ to 3′. |
Near the 5′ end of genes, there are promotor sequences that instruct messenger RNA (mRNA) to start transcription using the DNA antisense strand as a template. That is, a new RNA strand is made that is complementary in its base composition to the DNA antisense strand and thus is the same as the sense strand. Each DNA codon is thereby turned into a corresponding RNA codon.
The double helix uncoils, and the appropriate bases are added along the strand. RNA processing then occurs, cutting out (splicing) the introns, which do not code for anything, and rejoining the edges.
Translation then occurs. This refers to the new mRNA strands, which include only coding sequences that move out of the nucleus and into the cytoplasm where they bind to the ribosomes. The ribosomes assemble a polypeptide according to the recipe specified by the sequence of codons.
Various modifications may then occur before the chain is transported to its functional location within or outside of the cell.
DNA replicates itself in an analogous fashion. The two helical strands separate, and each is copied by a series of enzymes that position a complementary base opposite each base in the strand of DNA. One double helix thereby gives rise to two identical double helices.
Humans have approximately 2 m of DNA arranged into 46 chromosomes composed of 23 homologous pairs. This includes 44 autosomes, which are the same in males and females, plus either an XY in males (the Y chromosome determines testis development) or an XX in females (Fig. 3). Each member of a homologous pair carries corresponding, although not necessarily identical, genes in the same sequence. One member of each chromosome pair is inherited from the father, the other from the mother. For most of the cell cycle, each chromosome exists as a single chromatid. When a cell is committed to division, however, each chromatid is replicated. This is the form in which we usually see pictures of chromosomes: two identical sister chromatids joined at a centromere, making an “X” shape.
 Fig. 3. Normal human male XY karyotype showing 46 chromosomes. Sample was obtained from amniotic fluid. (Courtesy of Paul Fernhoff, MD, Emory University Genetics Laboratory) |
Each somatic cell has 46 chromosomes and divides by mitosis, which results in daughter cells with 46 chromosomes (Fig. 4). Mitosis gives rise to the body cells needed for growth and development. The normal sperm or ova, however, are the result of division by meiosis (Fig. 5) and have 23 chromosomes each, only one from each chromosome pair. In this manner, each parent passes on half of his or her genetic information to each child. Interestingly, the drive toward genetic diversity is so great that it can be seen even within a single sperm or egg. Each parent has two copies of every chromosome and passes on only one. However, the version of each chromosome that is passed on is not identical to either of the copies that the parent possesses. During meiosis, before the two copies of each chromosome separate to go to different eggs or sperm, the homologous chromosomes exchange genetic material in a process called crossing over (see Fig. 5B). At this stage, each homologue is made of two identical sister chromatids joined at the centromere. A crossover involves an exchange of genetic material between two chromatids on two different homologues.
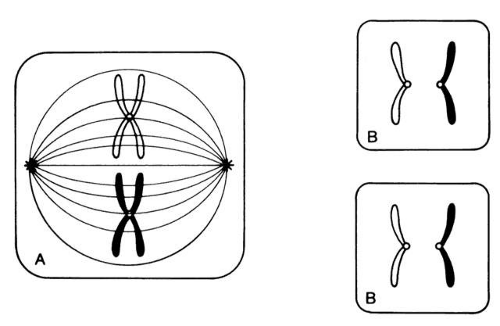 Fig. 4. Mitosis. Only one pair of homologous chromosomes is shown. A. Each chromosome has replicated to form two identical chromatids joined at the centromere. The color difference indicates that one chromosome is paternally derived and one maternally derived. Chromosomes line up in tandem at the equatorial plate before cell division. B. Daughter-cell chromosomes, shown as chromatids, are identical to those of the parent cell. |
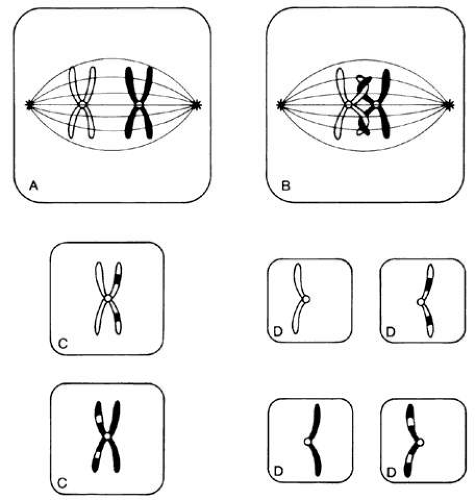 Fig. 5. Meiosis. In the first meiotic division, homologous chromosomes line up parallel to each other at the equatorial plane (A). Crossing over occurs between two of the sister chromatids (B), permitting the exchange of corresponding chromosomal segments. Reduction division results in daughter cells (C) that contain only one chromosome of each homologous pair. The second meiotic division produces daughter cells (D) with just one chromosome from each homologous pair, the haploid number. Here they are shown as chromatids, each one representing a different “arm” of the 4-armed homologous chromosome pair formed at the beginning of meiosis. |
For example, a person has one copy of chromosome 1 from her mother and another copy from her father. Assume that she has brown eyes and is esotropic. Assume hypothetically that an esotropia gene is present on the maternal chromosome 1 as well as a gene for brown eyes. On the paternal chromosome 1 are genes for straight eyes, color blue. We might expect that in meiosis, when one copy of each chromosome goes to each egg, that the resultant eggs would all have either the maternally derived chromosome 1 with brown eyes and esotropia or the paternal straight blue. In fact, however, crossing over exchanges material between this person’s two copies of chromosome 1, making unique versions to be passed on: very possibly esotropia with blue eyes or straight brown eyes. The chance that traits will be separated by crossovers increases as their genetic distance from each other increases.
Many crossovers may occur involving one, two, three, or four arms. In human male meiosis, an average of 55 crossovers occurs, with at least 1 crossover per homologous pair. Crossovers in females are even more frequent.10 Thus, the version of chromosome 1 passed on by a parent is actually an amalgam of different pieces from each of his or her two different chromosome 1 copies. This is important to understand because in some families, diseases caused by mutations in genes that are physically close together on the chromosome may occur together in some family members, whereas others may have only one trait or the other. It is also vital to understand crossing over because it plays an important role in understanding linkage analysis.
At conception, a sperm and ovum unite to form a zygote, which reestablishes the 46-chromosome complement. These chromosomes represent a random assortment of the genetic information present in each parent, in a unique combination that allows for advantageous new gene interactions in the offspring. Conversely, it may bring together genes that add to each other in a negative way, producing what we know as human malformation or disease. In addition, with such a complex process it is understandable that “mistakes” sometimes occur: chromosome pairs may not completely separate during meiosis, leading to extra genetic material in the offspring called trisomies or rearrangements called translocations. Trisomies occur with increased frequency as maternal age increases; mutations occur with increased frequency as paternal age increases. Pieces of DNA may be lost (deleted), or one or more bases may not pair up normally, changing a codon (mutations). These occurrences are not rare, and often result in early miscarriages. Although they are genetic in nature, they may or may not be inherited or heritable; it may be a chance occurrence without increased risk of recurrence.
A mutation is a stable, heritable alteration in DNA transmitted from parent to progeny.11 From an evolutionary standpoint, mutations are necessary to generate genetic diversity, which makes adaptation to differing environments possible. While some mutations cause disease, others contribute to success and survival of the fittest. Some mutations do both: when one of a person’s two homologous hemoglobin genes carries a sickle mutation, the person is resistant to malaria. Thus, this particular mutation protects persons who live in areas in which malaria is endemic, and it has been passed on to large numbers of progeny in some populations. If a person carries two copies of the sickle mutation, however, sickle cell anemia results, which is associated with high morbidity and mortality.
TAKING AN OPHTHALMIC FAMILY HISTORY
It is important to take a complete history, including pregnancy and birth, past medical history, medication use, and a detailed family history. For most patients, a few directed questions will yield the necessary information; for others, painstaking detail is required. Always remember that patients do not know which aspects of their family history will be important for the ophthalmologist to know, so if the right questions are not asked, valuable information will be lost.
Even a brief family history should be put in pedigree form using standard symbols for clarity (Fig. 6). Questioning should begin with names, ages, and genders of parents, siblings, and children of the proband (the person presenting for care). Initial questions should be general, and the type of questions to ask are common sense:
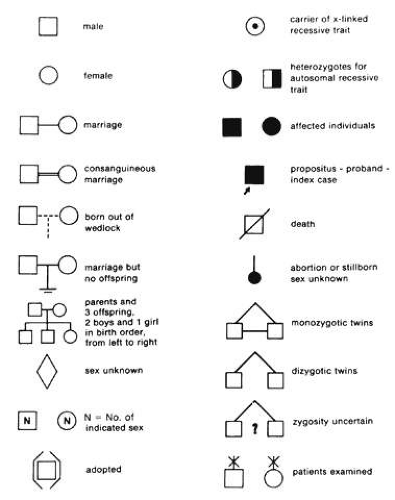 Fig. 6. The family history, or pedigree, is often of immeasurable value in reaching the correct diagnosis and ensuring appropriate genetic counseling. Above are the standard symbols used in drawing a pedigree. |
Does anyone else in your family have the same problem?
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


