Figure 9-1 A, Complete cryptophthalmos, both eyes. B, Incomplete cryptophthalmos of the right eye, with eyelid fused to cornea superonasally.
Cryptophthalmos occurs in both an isolated and a recessive syndromic form as Fraser syndrome. Patients with this syndrome may have a combination of acrofacial and urogenital malformations with or without cryptophthalmos. The disorder results from mutations in the FRAS1 gene located at 4q21, which encodes a putative extracellular matrix (ECM) protein.
Cryptophthalmos requires surgical intervention for cosmesis or relief of pain from absolute glaucoma. Pseudocryptophthalmos may benefit from fornix reconstruction using buccal mucosal and amniotic membrane grafts, with eyelid reconstruction to protect the corneas.
McGregor L, Makela V, Darling SM, et al. Fraser syndrome and mouse blebbed phenotype caused by mutations in FRAS1/Fras1 encoding a putative extracellular matrix protein. Nat Genet. 2003;34(2):203–208.
Stewart JM, David S, Seiff SR. Amniotic membrane graft in the surgical management of cryptophthalmos. Ophthal Plast Reconstr Surg. 2002;18(5):378–380.
Microphthalmos
Microphthalmos is a small, disorganized globe (Fig 9-2; Table 9-1). There is often an associated cystic outpouching of the posteroinferior sclera, likely due to a failure of the fetal fissure to close properly, and colobomatous defects of the iris, ciliary body, uvea, and optic nerve are often present.
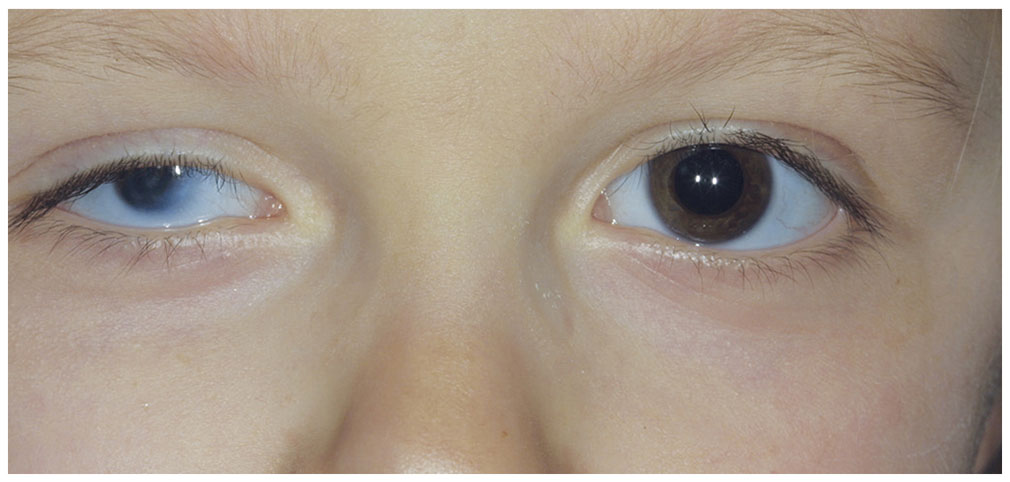
Figure 9-2 Microphthalmos, right eye. (Courtesy of Jeffrey Nerad, MD.)
Table 9-1
Multiple associations have been found with microphthalmos, including trisomies of almost every chromosome (typically, trisomy 13), maternal infections, and exposure to toxins and radiation. Most cases of nonsyndromic microphthalmos are sporadic, although autosomal dominant, autosomal recessive, and X-linked forms have been reported. Associated ocular abnormalities may include leukomas, anterior segment disorders, retinal dysplasia, colobomas, cysts, marked internal dysgenesis, persistent fetal vasculature (PFV), small orbit, ptosis, and blepharophimosis. Systemic associations are numerous, including intellectual disability and dwarfism.
Associated conditions should be sought and managed appropriately, and genetic counseling should be considered. A cosmetic shell or contact lens may be indicated in selected patients.
Ferda Percin E, Ploder LA, Yu JJ, et al. Human microphthalmia associated with mutations in the retinal homeobox gene CHX10. Nat Genet. 2000;25(4):397–401.
Li H, Wang JX, Wang CY, et al. Localization of a novel gene for congenital nonsyndromic simple microphthalmia to chromosome 2q11-14. Hum Genet. 2008;122(6):589–593.
Verma AS, FitzPatrick DR. Anophthalmia and microphthalmia. Orphanet J Rare Dis. 2007;2:47.
Nanophthalmos
Nanophthalmos is characterized by a small, functional eye with relatively normal internal organization and proportions. Patients have a high degree of hyperopia (7–15 diopters [D]) due to a short axial length (15–20 mm), and they also have a high lens-to-eye volume ratio that can lead to crowding of the anterior segment and angle-closure glaucoma. In addition, these patients have thickened sclera, steep corneal curvature, narrow palpebral fissures, and crowded anterior segments associated with angle-closure glaucoma. Many patients have strabismus. Choroidal effusions or hemorrhage has been frequently encountered during anterior segment surgery.
Nanophthalmos may be sporadic or hereditary, and both autosomal dominant and autosomal recessive inheritance patterns have been reported. One gene locus for the autosomal dominant form has been mapped to chromosome arm 11p. The recessive form of the disease is caused by a mutation in the gene encoding membrane-type frizzled protein (MFRP).
Laser iridotomy, sometimes combined with peripheral laser iridoplasty, may be effective treatment of the angle-closure component. Cataract surgery may be complicated by uveal effusion or hemorrhage and exudative retinal detachment, although advances in small-incision surgery have reduced the frequency of these complications. Extremely high intraocular lens powers are required to achieve emmetropia.
Faucher A, Hasanee K, Rootman DS. Phacoemulsification and intraocular lens implantation in nanophthalmic eyes: report of a medium-size series. J Cataract Refract Surg. 2002;28(5):837–842.
Traboulsi EI, ed. Genetic Diseases of the Eye. 2nd ed. Cary, NC: Oxford University Press; 2011.
Blue Sclera
The striking clinical picture of blue sclera is related to generalized scleral thinning, with increased visibility of the underlying uvea. This anomaly must be distinguished from the slate-gray appearance of ocular melanosis bulbi and from acquired causes of scleral thinning such as rheumatoid arthritis or staining from minocycline treatment.
Two syndromes associated with blue sclera are osteogenesis imperfecta type I and Ehlers-Danlos syndrome type VI (see Table 9-1). Osteogenesis imperfecta type I is a dominantly inherited generalized connective tissue disorder characterized mainly by bone fragility, in addition to blue sclerae. Ehlers-Danlos syndrome type VI is a rare syndrome with autosomal recessive inheritance. In addition to blue sclera, keratoglobus, and keratoconus, it is characterized by joint hyperextensibility, severe kyphoscoliosis, cardiac anomalies, and skin abnormalities of easy bruisability, abnormal scarring, and soft distensibility. See Chapter 11 for further discussion of Ehlers-Danlos syndrome. These syndromes may share similar manifestations of fractures from minor trauma in childhood, kyphoscoliosis, joint extensibility, and elastic skin. Decreased hearing and tinnitus may also occur.
Regular hearing evaluations after adolescence are recommended. Oral bisphosphonate therapy may be specifically indicated for these patients. Postmenopausal women should engage in a long-term physical therapy program to strengthen the paraspinal muscles. Estrogen and progesterone replacement and adequate calcium and vitamin D intake are indicated. Fractures are treated with standard methods. Future therapies may include stem cell transplantation and gene therapy.
Developmental Anomalies of the Anterior Segment
See Table 9-1 for a summary of developmental anomalies of the anterior segment.
Anomalies of Size and Shape of the Cornea
Microcornea
Microcornea refers to a clear cornea of normal thickness whose diameter is less than 10 mm (or <9 mm in a newborn). If the whole anterior segment is small, the term anterior microphthalmos applies. If the entire eye is small and malformed, the term microphthalmos is used in contrast to nanophthalmos, in which the eye is small but otherwise relatively normal.
The cause is unknown and may be related to fetal arrest of growth of the cornea in the fifth month. Alternatively, it may be related to overgrowth of the anterior tips of the optic cup, which leaves less space for the cornea to develop.
Microcornea may be transmitted as an autosomal dominant (most commonly) or recessive trait with equal sex predilection. Because their corneas are relatively flat, patients with microcornea are usually hyperopic and have a higher incidence of angle-closure glaucoma. Of patients who do not develop angle-closure glaucoma, 20% develop open-angle glaucoma later in life. Important ocular anomalies often associated with microcornea include PFV, congenital cataracts, anterior segment dysgenesis, and optic nerve hypoplasia. Significant systemic associations include myotonic dystrophy, fetal alcohol syndrome, achondroplasia, and Ehlers-Danlos syndrome.
If microcornea occurs as an isolated finding, the patient has an excellent visual prognosis with spectacles to treat the hyperopia resulting from the flat cornea. Concurrent ocular pathologic conditions such as cataract, PFV, and glaucoma may require treatment following the usual procedures for those conditions.
Megalocornea
Megalocornea is a bilateral, nonprogressive corneal enlargement with an X-linked recessive inheritance pattern (see Table 9-1). Rare cases of autosomal recessive inheritance have been reported. Affected persons have histologically normal corneas measuring 13.0–16.5 mm in diameter (Fig 9-3). Males are more typically affected, but heterozygous women may demonstrate a slight increase in corneal diameter.
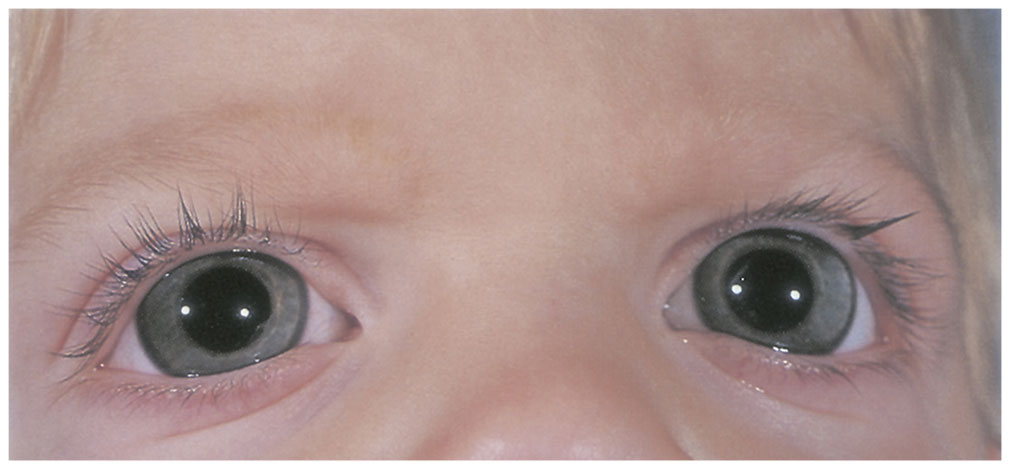
Figure 9-3 Megalocornea.
The etiology may be related to failure of the optic cup to grow and of its anterior tips to close, leaving a larger space for the cornea to fill. Alternatively, megalocornea may represent arrested buphthalmos and exaggerated growth of the cornea in relation to the rest of the eye. An abnormality in collagen production is suggested by the association of megalocornea with systemic disorders of collagen synthesis (eg, Marfan syndrome).
Megalocornea may be associated with iris translucency (diaphany), miosis, goniodysgenesis, cataract, ectopia lentis, arcus juvenilis, and glaucoma (but not congenital glaucoma). Systemic associations include craniosynostosis, frontal bossing, hypertelorism, facial anomalies, dwarfism, facial hemiatrophy, intellectual disability, hypotonia, Down syndrome, Marfan syndrome, Alport syndrome, osteogenesis imperfecta, mucolipidosis type II, or occasionally other genetic syndromes.
Congenital glaucoma must be ruled out by intraocular pressure measurement and careful biomicroscopy. Ultrasonography may be of value in determining the short vitreous length, deep lens and iris position, and normal axial length that distinguish megalocornea from buphthalmos caused by congenital glaucoma. Myopia and with-the-rule astigmatism are managed as in unaffected patients. Care must be taken during cataract surgery to implant the intraocular lens into the lens capsular bag. Standard-sized posterior chamber lenses are typically too short to be fixated in the ciliary sulcus, and anterior chamber lenses are similarly problematic in the enlarged anterior chamber.
Mackey DA, Buttery RG, Wise GM, Denton MJ. Description of X-linked megalocornea with identification of the gene locus. Arch Ophthalmol. 1991;109(6):829–833.
Traboulsi EI, ed. Genetic Diseases of the Eye. 2nd ed. Cary, NC: Oxford University Press; 2011.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


