Causes of extrinsic airway obstruction
#Oral
##Epignathus
##Epulus
##Micrognathia
##Parotid tumor
#Cervical
##Cervical teratoma
##Lymphangioma
##Congenital goiter
##Solid thyroid tumor
##Cystic anomaly of thyroid
##Branchial cleft cyst
##Hamartoma
##Choristoma
##Hemangioma
##Lipoma
##Anterior neural tube defects
##Twins sac of a blighted ovum
#Thoarcic
##Bronchogenic cyst
##Mediastinal teratoma
##Pericardial teratoma
##Congenital pulmonary airway malformation
#Causes of intrinsic airway obstruction
##Laryngeal atresia
##Laryngeal cyst
##Laryngeal web
##Laryngeal stenosi
##Tracheal atresia
##Tracheal stenosis
##Bronchial atresia
##Bronchial web
Epignathus
Epignathus is an oropharyngeal teratoma arising from the sphenoid bone, palate, or pharynx. The cells of origin are likely pluripotent stem cells derived from cells in Rathke’s pouch. The tumor may vary considerably in size but tumors that fill the oral cavity and protrude through the mouth are common, often leading to obstruction of the upper airway. Without prompt intervention at time of delivery, most newborn infants will succumb to airway obstruction and die soon after birth [1]. Epignathus is fortunately a rare tumor with an estimated incidence range from 1:35,000 to 1:200,000 [2]. Large epignathus can present early in the second trimester with polyhydramnios and complete airway obstruction at the level of the oro-pharynx [3]. Polyhydramnios can be severe in this setting and may require serial reduction to avoid premature labor [4]. In addition to ultrasound, an ultra-fast MRI scan can provide important anatomic information about the mass, confirm the origin of the tumor, vascular supply, and secondary effects of the mass on the mandible and other adjacent structures (Fig. 1).
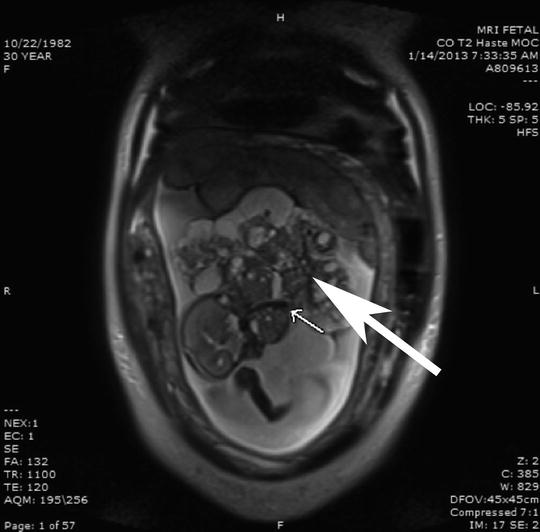
Fig. 1
Fetal MRI in sagittal section of a 25-week gestation fetus with very large exophytic epignathus with mass larger than the fetus but arising from the hard palate in the fetal mouth. The arrow points to a large vessel creating a “flow void” consistent with the major systemic blood supply to the epignathus
Management of the fetus with epignathus depends upon the size of the mass and the degree of airway compromise, gestational age at diagnosis, and the potential physiologic derangements caused by the tumor [4]. In many cases, the epignathus is small and does not obstruct the fetal airway and can be readily managed at the time of delivery by conventional means. In contrast, at the other end of the spectrum, epignathus can grow very rapidly and to large proportions exophytically. This leads to increased blood flow through the tumor, high output congestive heart failure, and fetal hydrops. In addition, these large fetal epignathus can create such large metabolic demands on the fetus that it induces fetal growth arrest. Both of these fetal presentations warrant considering fetal surgery in order to afford the fetus any chance of survival. Despite its large size, the mass invariably arises from a very narrow pedicle at the fetal palate. It may be approached by open fetal surgery (Fig. 2), or if there is a single systemic arterial feeding vessel (usually derived from the palatal arteries), by interstitial laser coagulation of the feeding vessel. If the fetus is at a gestational age ≥ 28 weeks gestation consideration should be given to ex utero intrapartum treatment (EXIT) to resect the mass and secure the airway prior to delivery. See section on EXIT procedure below.
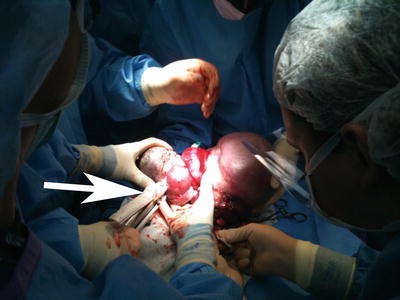
Fig. 2
Intraoperative photograph taken during EXIT-to-airway for giant epignathus. The arrow is pointing at a thick tissue stapling device which is being applied to take the mass off at the mouth. The size of the mass makes it near impossible to resect the mass without first debulking. This also allows the airway to be secured either by nasotracheal intubation or tracheostomy tube placement
Congenital Epulis
Congenital epulis is also known as Neumann’s tumor or gingival granular cell tumor (GGCT) of the newborn [5, 6]. The tumor arises from the mucosa of the gingiva, most commonly from the anterior part of the maxillary or mandibular ridge. Similar to epignathus, epulis of the newborn is an oral mass (or masses), which protrudes through the mouth and can obstruct the airway (Fig. 3). Congenital epulis has a female preponderance of 8:1 and can vary in size from a few millimeters to as large as 7.5 cm. The GGCT seen in newborns has different histologic features than those originating elsewhere [7].
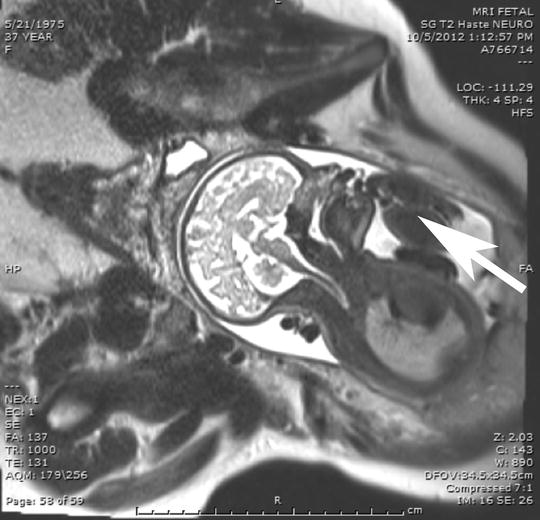
Fig. 3
Not every oral mass requires fetal MRI to demonstrate an oral mass. The image shows a fetus with an epulis, the arrow is pointing at the mass, which was not sufficiently large to compromise the airway and did not require the EXIT strategy
Epulis in the newborn has been reported to spontaneously regress after birth, suggesting that its growth is dependent on maternal hormonal milieu associated with pregnancy [8]. If sufficiently large, these tumors can compromise the upper airway, but these cases are rare. Intervention at birth may be necessary, depending on the size and likelihood of airway obstruction [4]. Epulis usually originate from a pedicled stalk and can often be easily resected before establishing an airway in the controlled environment of an EXIT-to-airway procedure [4]. If the diagnosis has not been made prenatally, and the epulis is large, establishing an airway may be difficult and may require emergency resection or tracheostomy.
Micrognathia
Severe underdevelopment of the fetal mandible can result in Pierre-Robin sequence (PRS) and is associated with micro-retrognathia and glossoptosis, causing airway obstruction. A small mandible characterizes PRS, and it is typically accompanied by a U-shaped cleft palate [9]. PRS occurs in 1:8,500–1:14,000 births. It can be seen alone or as part of a syndromic presentation [10]. About 40 % of PRS cases occur in isolation and 60 % present with an associated syndrome, most commonly Stickler, Nagger, and velocardiofacial syndromes (see Table 3) [10]. Arthrogryposis multiplex is a rare congenital disease characterized by nonprogressive multiple joint contractures at birth. It is also associated with micrognathia, a small mandible, and cleft palate [11].
Identifying micrognathia with or without associated polyhydramnios or cleft palate leads to the prenatal diagnosis of PRS (Fig. 4). In a fetus with PRS, the severity of micrognathia will determine the likelihood of airway obstruction. Airway obstruction is more likely to be present if it is associated with evidence of aerodigestive obstruction, as indicated by polyhydramnios, glossoptosis, or the absence of fluid in the stomach [12]. Ultrasound is most commonly used to assess the presence and severity of micrognathia, which subjectively may be evident on sagittal images of the fetal head. The jaw index (Fig. 5), described by [47] offers a more objective measure of micrognathia and is defined by the ratio of the anterior–posterior length of the mandible divided by the biparietal diameter multiplied by 100. When the jaw index is less than the fifth percentile and is associated with evidence of aerodigestive obstruction, such as glossoptosis, polyhydramnios, or absent stomach bubble, [48] have suggested that an EXIT-to-airway is indicated. In these severe cases of micro-retrognathia a tracheostomy is necessary to establish an airway, and is most safely and effectively achieved as part of EXIT-to-airway procedure.
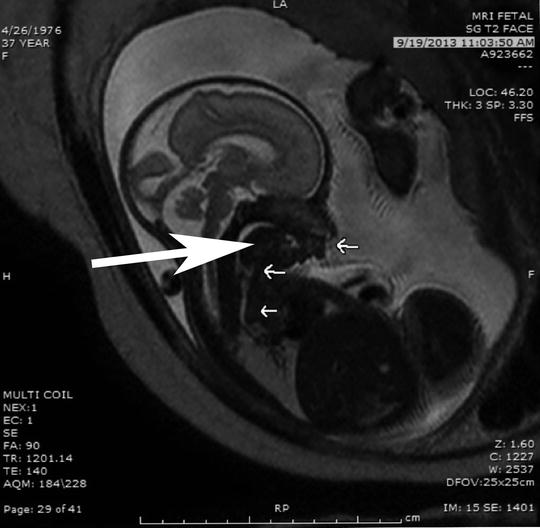
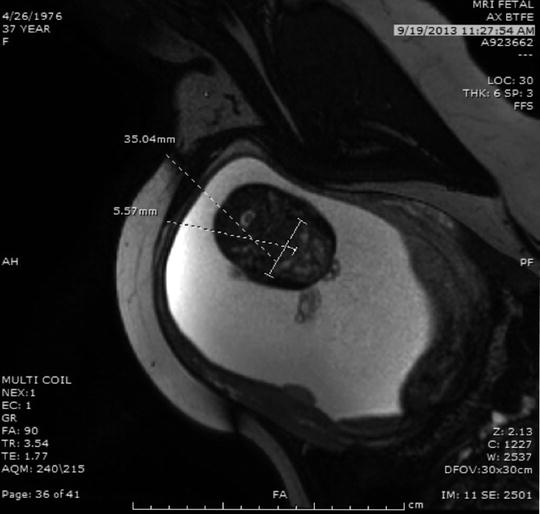
Fig. 4
Fetal MRI image in profile demonstrating severe micrognathia (small arrows). This fetus had micrognathia as part of Nager syndrome and had a jaw index of less than the fifth percentile with associated glossoptosis, the arrow is pointing at the tongue, as can be seen by the tongue displaced posteriorly. There was also absence of fluid in the stomach by ultrasound indicating inability to swallow amniotic fluid due to the glossoptosis. This is consistent with the presence of a critical airway meeting criteria for EXIT-to-airway strategy
Fig. 5
MRI image demonstrating the technique to obtain the jaw index. A line is drawn from the angles of the mandible and an orthogonal line is drawn to the mentum of the mandible. This later measurement is then divided by the biparietal diameter to yield a ratio that is multiplied by 100. A value less than the fifth percentile (jaw index < 24) would indicate fetuses at highest risk for airway compromise and when associated with glossoptosis and absence of fluid in the stomach would be an indication for EXIT-to-airway
Cervical Lymphangioma (Vascular Malformation)
Lymphangioma is the former term still commonly used for lymphatic vascular malformations. These benign tumors can grow to large proportions and significantly compromise the fetal airway (Fig. 6). They are often present at birth and usually clinically evident as a soft tissue mass of the neck. Vascular malformations can also present in the axilla, thorax, and lower extremities, although this occurs less frequently [13]. When a vascular malformation of the neck is large and comprised of macrocystic components, it has commonly been referred to as a cystic hygroma.
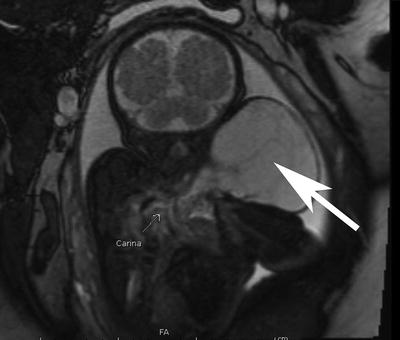
Fig. 6
The coronal section of a fetal MRI demonstrate the appearance of a large cervical lymphatic vascular malformation, an arrow is pointing at the malformation, it is extending from the fetal left neck and causes minimal compression of the midline airway. The mass extends into the upper left chest but does not cause significant airway compromise. This vascular malformation was not particularly vascular and would not be expected to critically compromise the airway at delivery. However, rapid enlargement post-delivery is not uncommon. Conventional delivery followed by intubation would be the safest way to prevent development of airway compromise until the vascular malformation can be fully evaluated postnatally
The lymphatic system develops at the end of the fifth week of gestation as a result of six primary lymph sacs sprouting in the neck, iliac region, and retroperitoneum. One theory on the formation of lymphatic vascular malformations is that they are developmental defects secondary to sequestration of lymphatic tissue in early embryonic life [14]. Cervical lymphatic vascular malformations are believed to result from a failure of cervical lymph sacs to join the lymphatic channels. As a result, endothelial-lined spaces secrete lymph-like fluid, which leads to local distention and gradual enlargement of cysts. Over time the walls of these cysts thicken, and connective tissue septae separate large cysts [15].
The prevalence of prenatally diagnosed lymphatic vascular malformations has been estimated to be 1 in 1,775 live births [16]. The incidence may be as high as 1 in 300 among spontaneously aborted fetuses [17]. The mortality of lymphatic vascular malformations diagnosed prior to 20 weeks is high, due to associated chromosomal anomalies, syndromes, and non-immune hydrops [18, 19]. The incidence of chromosomal abnormalities may be as high as 60 %, and additional structural abnormalities are common. Lymphatic vascular malformations diagnosed prior to 20 weeks may be associated with Turner syndrome, oligohydramnios, single vessel umbilical cord, Noonan syndrome, fetal alcohol syndrome, Fryn syndrome, trisomies 18 and 21, and hydrops fetalis [18–22]. Lymphatic vascular malformations diagnosed early in gestation are typically located in the posterior triangle of the neck and are associated with lymphangiomatosis. In contrast, isolated cervical lymphatic vascular malformations usually present in the third trimester or after birth, often following a normal ultrasound earlier in pregnancy [18]. Thus, the etiology for early and late appearing lymphatic vascular malformations is suspected to be different.
The natural history of cervical lymphatic vascular malformations is largely determined by the gestational age at the time of diagnosis. Diagnosis early in pregnancy, before 20 weeks gestation, is associated with multiple anomalies, karyotype anomalies, and a high rate of spontaneous abortion with mortality exceeding 90 %. If a lymphatic vascular malformation is diagnosed later in pregnancy, after 20 weeks gestation, no other anomalies are found on prenatal imaging, and the karyotype analysis is normal, the outcome is almost uniformly favorable, with the exception of the impact on the fetal airway. In this subset, careful evaluation of the airway is necessary to assure a safe transition to postnatal life, as this transition often results in rapid enlargement of the vascular malformation. Because most of these later gestation age lymphatic malformations arise from the lateral neck there may be impingement or displacement of the fetal airway (Fig. 6). In many cases conventional management of the airway is possible. However, even in cases in which intubation is not initially deemed necessary, it is important to consider intubation to secure the airway, because of the rapid enlargement of the vascular malformation following delivery. In cases where the size of the lymphatic vascular malformation is large, crossing the midline, and severely compromising the fetal airway, it may be difficult to distinguish from cervical teratoma, and warrants serious consideration of an EXIT-to-airway.
Cervical Teratoma
Teratomas are germ cell tumors composed of tissue foreign to their anatomical location. Teratomas contain tissue from all three germ layers and are most commonly located in the sacrococcygeal region, but can occur in the abdomen, chest, neck, or intracranially. The incidence of teratoma in newborn infants is approximately 1:40,000 [23]. Teratomas of the neck are less common, comprising somewhere between <5 and 13 % of the total number of teratomas in newborns [24, 25]. Cervical teratomas are more often diagnosed prenatally and less often contain yolk sac components [24], which are considered to be a histologic feature suggesting malignancy. There is no gender or race predilection [24]. The most common tissue present in cervical teratomas is neural tissue. However, cervical teratomas contain thyroid tissue in 30–40 % of cases—it is unknown if this represents involvement of the thyroid gland or ectopic tissue [25].
Cervical teratomas are usually bulky, often 5–12 cm in size [26]. They often extend from the mastoid process and body of the mandible to the clavicle and sternal notch, displacing the ear superiorly. Mandibular hypoplasia can occur as a result of a mass effect on the mandible, often with compression of the marginal mandibular branch of the facial nerve and associated droop of the corner of the mouth. The posterior border of cervical teratomas often reaches the anterior border of the trapezius muscle. Cervical teratomas result in polyhydramnios in about 40 % of cases, which is the result of esophageal compression by the mass [27]. An empty stomach indicates that the polyhydramnios present is secondary to a mass effect on the esophagus and can be the initial finding leading clinicians to the diagnosis of a cervical teratoma [28]. Cervical teratomas may also have calcifications within the mass and compared to lymphatic vascular malformations tend to be more solid.
Lymphatic vascular malformations and cervical teratomas can be difficult to differentiate on prenatal ultrasound [26]. Cervical teratomas usually have a well-defined border when compared to the multi-loculated, cystic appearance of a cervical lymphatic vascular malformation. Cervical teratomas are often bulkier tumors and located in the anterior neck (Fig. 7). Alpha feto protein (AFP) is not useful in differentiating lymphatic vascular malformations from cervical teratoma, as less than 30 % of cases of cervical teratomas have an elevated AFP [29].
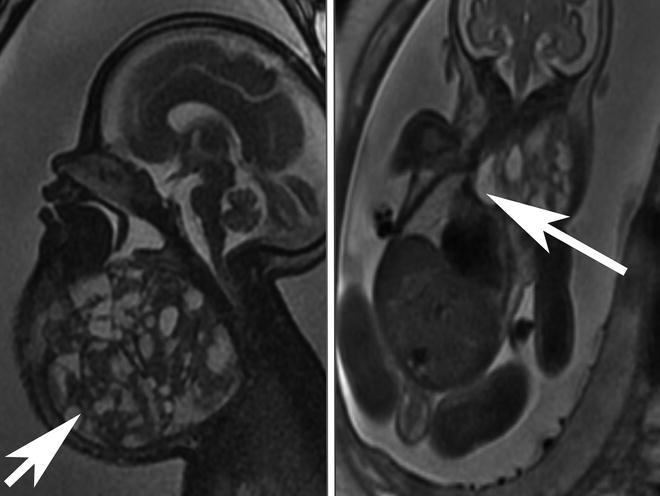
Fig. 7
The panel on the left shows a sagittal section of a fetal MRI showing a very large cervical teratoma, the arrow is pointing at the mass, with both cystic and solid components. Calcifications are difficult to see on MRI but were present on ultrasound. The coronal section seen in the panel on the right shows a MRI image of the same patient demonstrating extension of the teratoma into the mediastinum through the thoracic inlet, the arrow is pointing at the mass. This mass resulted in a SVC syndrome with swelling of scalp and facial edema. The compromise of the cervical airway and the SVC syndrome with associated venous hypertension made EXIT-to-airway through the neck unsafe. In this case a transthoracic retrograde intubation EXIT (TRI-EXIT) allowed an airway to be secured by median sternotomy during the EXIT with retrograde passage of ETT changer over which an ETT was placed and the trachea repaired
The majority of cervical teratomas are benign, but the natural history is not well characterized. One case series suggests metastasis occurs in 14 % of cervical teratomas in the form of immature neuroglial elements in regional lymph nodes, but found that only 1.4 % of cervical teratomas had yolk sac components [24]. Many infants have remained free of disease long term after resection, even in the setting of metastatic lesions to regional lymph nodes. This could indicate that the tumor biology behaves more like a benign tumor even in the setting of regional spread of disease [30].
Cervical teratomas are predisposed to develop into large neck masses in utero, which commonly compromise the fetal airway, and often require management by EXIT-to airway at a mean gestational age of 34 weeks [31].
Intra-thoracic Extrinsic Airway Obstruction
Bronchogenic Cyst
Bronchogenic cysts can cause airway obstruction in the fetus and in the neonate immediately after birth. The degree and location of obstruction can be quite variable and this information will inform decisions in regards to delivery and intervention.
If the bronchogenic cyst arises from the trachea or one of the main-stem bronchi, it can cause complete obstruction of the airway. In utero this can cause “hyperinflation” of the lung distal to the obstruction and malacia of the airway at the area of obstruction (Fig. 8). This can result in a ball-valve effect with fluid trapping in the lung before birth and air trapping at delivery. If one of the main-stem bronchi is affected this can result in mediastinal shift and compression of the opposite lung with resultant respiratory collapse. If the trachea is affected, both lungs could be compromised due to surfactant deficiency and inability to clear secretions. Airway obstruction in utero results in less type II pneumocytes and surfactant deficiency.
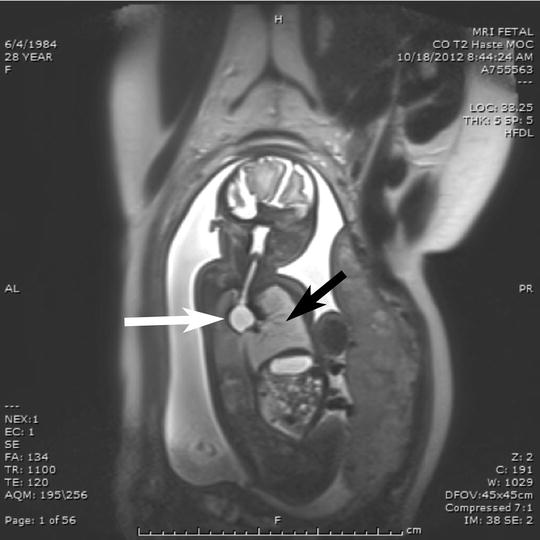
Fig. 8




Fetal MRI demonstrating a large bronchogenic cyst, a white arrow is pointing at the cyst, compressing the left main-stem bronchus, the black arrow is pointing at the “hyperinflated” left lung. By the time of delivery, the left lung was very enlarged and herniated across the midline into the right hemithorax. EXIT-to-resection was performed allowing time to perform a right thoracotomy and resect the bronchogenic cyst. In addition, bronchoscopy was performed to evaluate the compressed trachea for malacia and to administer surfactant protein selectively to the left lung, which due to obstruction was surfactant deficient from type II pneumocyte down regulation
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
