(1)
Department of Ophthalmology and Visual Sciences, University of Iowa Hospitals and Clinics, Iowa City, IA, USA
Arterial hypertension is a common systemic disease. In the middle-aged and elderly population, there is a trend toward a rise in blood pressure (BP) which often becomes steeper with age [1]. Systemic mortality and morbidity are markedly higher in hypertensives than in normotensives; for example, hypertensives are seven times as likely to develop stroke as normotensives, four times as likely to have congestive heart failure, three times as likely to have coronary artery disease, and twice as likely to have peripheral arterial disease; and the hypertensive’s risk of total and cardiovascular mortality is twice that of the normotensive [2].
As regards the eyes, it is evident from the discussion of the pathogeneses of various ocular vascular occlusive disorders in this book that arterial hypertension plays an important role in their development. Not only that but also ophthalmic manifestations may be the initial clinical presentation of arterial hypertension, particularly of malignant arterial hypertension. Richard Bright [3], an English physician, was the first to describe the association of renal disease with visual disturbances in 1836. In 1859, Liebreich [4] first described fundus changes in malignant arterial hypertension. Hypertensive fundus changes constitute a very important part of the syndrome of malignant arterial hypertension, as discussed below. A huge amount of literature has accumulated on the hypertensive fundus changes, drawn from both clinical and experimental studies. While clinical observations and studies are extremely helpful in understanding a disease process, they have some serious limitations. Over recent years, several advances have helped greatly to improve our understanding of the field, particularly about the pathology and pathogenesis of the various hypertensive fundus lesions. These include (a) a better understanding of basic properties of the ocular and optic nerve head vascular beds and their response to malignant hypertension, (b) ability to produce malignant arterial hypertension experimentally in primates [5, 6], (c) the advent of fluorescein fundus angiography and its application in hypertensive fundus changes, and (d) other modern investigative techniques.
I investigated fundus lesions in malignant arterial hypertension by experimental studies [6–18] in rhesus monkeys and also clinically. The discussion in this chapter is mostly based on those studies, with a brief review of the relevant literature.
First, for a proper comprehension of the mechanism of production of the various fundus lesions and their clinical pattern seen with malignant arterial hypertension, it is essential to understand some very basic anatomic and physiologic properties of the retinal, choroidal, and optic nerve head vascular beds and their response to malignant arterial hypertension. Following is a brief account of these properties.
Anatomic and Physiologic Properties of the Retinal, Choroidal, and Optic Nerve Head Vascular Beds
Ocular Blood Flow
This is discussed at length in Chap. 10. The following related topics need brief discussion.
Autoregulation of Blood Flow: This is discussed in detail in Chap. 10. Autoregulation may be disrupted by alterations in blood pressure (BP) produced by a variety of local and systemic causes, including the following.
Rise or Fall of Perfusion Pressure Beyond the Critical Autoregulatory Range
Autoregulation operates only over a critical range of perfusion pressure; with a rise or fall of perfusion pressure beyond the critical range, the autoregulation becomes ineffective and breaks down [19] (Fig. 23.1). Thus, autoregulation does not protect the tissues all the time. If the perfusion pressure rapidly goes above the autoregulation range (as in malignant hypertension), it damages the tissue; and if it falls below the range (as in marked arterial hypotension), the tissue is subjected to a risk of ischemia.
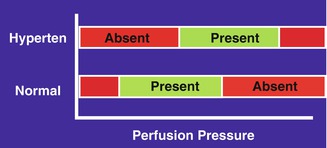
Fig. 23.1
A diagrammatic representation of blood flow autoregulation range at different perfusion pressures in normal persons and in hypertensives (Hyperten). Red autoregulation absent, green autoregulation present (Reproduced from Hayreh [19])
With chronic malignant hypertension and essential arterial hypertension, the autoregulation adjusts itself to higher than normal levels as a compensatory mechanism [20, 21]. In this, the range of autoregulation shifts to higher levels to adapt to high BP (Fig. 23.1). Although such an adjustment improves the patient’s tolerance to high BP, it makes that person correspondingly less tolerant to low BP, so that a level of BP that would normally be quite safe becomes dangerous. Under such circumstances, a large, sudden fall of BP in a hypertensive (spontaneously, as occurs during sleep, or because of overtreatment with antihypertensive drugs) can cause the perfusion pressure to fall below the autoregulatory range and result in systemic and/or ocular vascular accidents; these lesions may erroneously be attributed to hypertension, because neither the physician nor the patient may be aware of episodes of marked hypotension occurring during sleep (nocturnal hypotension) in hypertensives [22]. Figure 23.1 is a diagrammatic representation of blood flow autoregulation range at different perfusion pressures in normal persons (“normal”) and in hypertensives (“hyperten”). The green area represents the presence of autoregulation and the red area its absence.
Changes in Size of the Lumen of the Precapillary Arterioles
The main factor normally regulating the blood flow is thought to be the size of the lumen of the precapillary arterioles. Arteriolar changes in hypertension are well known. These include vasospasm, vasodilatation, arteriolosclerosis, drug-induced vasoconstriction or dilatation, or vasoconstriction caused by angiotensin (leaked into the optic nerve head and choroid – see below). All these arteriolar changes can interfere with autoregulation in arterial hypertension.
Changes in Vascular Endothelial Function
These occur early in the course of vascular diseases. In hypertension, morphologic and functional alterations occur in endothelial cells and basal formation of nitric oxide (a vasodilator) is reduced [23]. Normal vascular tone is determined by the balance between the vasodilators and vasoconstrictors. Reduced formation of dilating agents by the endothelium may lead to unopposed action of endothelin (a powerful vasoconstrictor), causing vasoconstriction and interfering with autoregulation (Fig. 23.2). Experimental data suggest that endothelial dysfunction develops as BP increases, and the dysfunction is related to the level of the BP. Therefore, endothelial dysfunction may contribute to hypertensive vascular complications and accidents [23]. There is evidence to suggest that damage to vascular endothelium (associated with abnormalities in production of vascular endothelium-derived vasoactive agents) also occurs in arteriosclerosis, atherosclerosis, hypercholesterolemia, aging, diabetes mellitus, ischemia, and from other causes so far unknown [23–25]; these conditions are often associated with arterial hypertension and would aggravate the situation.
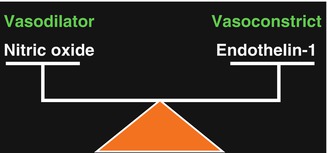
Fig. 23.2
A diagrammatic representation of balancing role of nitric oxide and endothelin-1 in modulating local vascular tone
Blood-Ocular Barrier
This is discussed at length in Chap. 9. Briefly, the presence of the blood-retinal barrier in the retina is well established. The choroid and optic nerve head, by contrast, do not possess blood-ocular barriers. With a severe rise of BP (i.e., above the level of autoregulation), the autoregulation breaks down, resulting in focal or generalized dilatation of retinal arterioles [7]. Morphologic studies have revealed discontinuity of the endothelial cell layer or interendothelial separation [5]. These changes result in the failure of the blood-retinal barrier and increased permeability. The barrier at the level of retinal pigment epithelium (RPE) also breaks down when the retinal pigment epithelial cells are destroyed or subjected to ischemia, as in hypertensive choroidopathy [10, 11]. Since the retinal tissue itself has no barrier in its stroma, fluid may be able to diffuse from one part to the adjacent areas [10]. Malignant arterial hypertension may derange one or both of the blood-retinal barriers. It is well established that in contrast to the blood vessels in the retina, choriocapillaris possesses no blood-ocular barrier because its endothelium shows the presence of numerous fenestrations. These make the choriocapillaris freely permeable [26–28], with a permeability to plasma proteins about five times that of the kidney; the choriocapillaris’ permeability to low-molecular-weight substances is even higher than that to macromolecules [29]. While the blood vessels in the optic nerve head, by virtue of tight cell junctions, have a blood-optic nerve barrier, it is important to remember that the optic nerve head itself does not possess a blood-ocular barrier. This is because the border tissue of Elschnig (separating the peripapillary choroid and optic nerve head – (Fig. 23.3) [30] allows choroidal interstitial tissue fluid to leak into the optic nerve head from the peripapillary choroid [8].
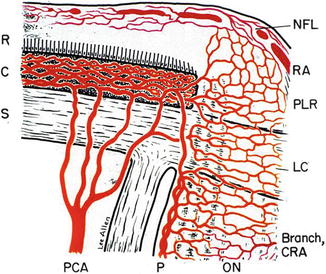
Fig. 23.3
Schematic representation of blood supply of the optic nerve head (Reproduced from Hayreh [30].) Abbreviations used: C choroid, CRA central retinal artery, LC lamina cribrosa, NFL surface nerve fiber layer of the disc, ON optic nerve, P pia, PCA posterior ciliary artery, PLR prelaminar region, R retina, RA retinal arteriole, S sclera
Thus, the retinal, choroidal, and optic nerve head blood vessels, because of their different anatomic and physiologic properties, respond differently to malignant arterial hypertension, thereby making hypertensive retinopathy, hypertensive choroidopathy, and hypertensive optic neuropathy three distinct and unrelated manifestations of malignant hypertension. This is a fact not fully appreciated in the past.
Pathophysiology of Malignant Arterial Hypertension
To understand the pathogenesis of various fundus lesions in malignant arterial hypertension, it is also essential to have some idea of the latter’s pathophysiology. This is still not fully understood. Arterial hypertension is considered a multifactorial disease secondary to the interaction of many abnormalities, including the following [31]:
1.
Abnormalities in cell membrane, resulting in defective membrane control over intracellular calcium concentration.
2.
Abnormalities of calcium metabolism, causing a rise in cytoplasmic calcium, which in turn causes increased tone of the arteriolar smooth muscle.
3.
Abnormalities of sodium metabolism, from inherited difficulty in the kidney’s ability to eliminate sodium.
4.
Abnormalities of potassium metabolism affect biosynthesis of aldosterone and renin release.
5.
Abnormalities of the central nervous system, including abnormal release of humoral factors (e.g., natriuretic factor, vasopressin) which increase sympathetic discharge and increase neurogenic vasomotor tone.
6.
Abnormalities of prostaglandins.
7.
Abnormalities of vascular endothelium-derived vasoactive agents (e.g., endothelin-1 [a powerful vasoconstrictor] and nitric oxide [a vasodilator]).
8.
Genetic effects on BP are polygenic in nature, and play an important role in the development of arterial hypertension.
9.
Abnormalities of the renin-angiotensin-aldosterone system; available evidence strongly indicates that abnormalities of this system play an important role in the development and maintenance of renovascular malignant arterial hypertension.
By some ill-understood mechanism(s), the levels of circulating endogenous vasoconstrictor agents (e.g., angiotensin II, epinephrine, vasopressin, and endothelin-1) increase. Kincaid-Smith [32] proposed the following schema for the various vascular changes seen in malignant arterial hypertension: increase of endogenous vasoconstrictor agents → “sausage string” effect in arteries → turbulence and endothelial separation and damage → platelet deposition on endothelium → release of thromboxane, serotonin, histamine → microangiopathic hemolytic anemia and intravascular coagulation → further platelet and fibrin deposition → mitogenic and migration factors released by platelet aggregation → myointimal proliferation and organization of thrombi within vessels → renal ischemia → increase in vasoactive agents; thus, a vicious circle is set up in malignant hypertension. Probably similar vascular changes in the ocular vessels may be developing and playing an important role in the development of hypertensive retinal, choroidal, and optic nerve head lesions.
Terminology Used for Hypertensive Fundus Changes
In 1859, Liebreich [4] first described fundus changes in malignant arterial hypertension under the title of “albuminuric retinitis.” Since then a variety of terms have been suggested to describe these changes [13]. “Hypertensive retinopathy” has been used as a universal term for all the fundus changes. However, in view of the above description of the basic properties of the retinal, choroidal, and optic nerve head vascular beds, it is clear that each one responds very differently to malignant arterial hypertension. Thus, pathogenetically and clinically, fundus changes in malignant arterial hypertension fall into three very distinct categories: (1) hypertensive retinopathy, (2) hypertensive choroidopathy, and (3) hypertensive optic neuropathy. This is evident from the following discussion of the three clinical entities seen in experimental studies.
Experimental Studies on Fundus Changes in Malignant Arterial Hypertension
Goldblatt and coworkers [33] produced renovascular arterial hypertension in 1934. This method has been used extensively ever since, with some modification, to produce experimental hypertension in order to study different aspects of this disease. Since 1937, several studies have investigated ophthalmic findings by producing experimental arterial hypertension in different animal species, which I briefly summarized elsewhere [6]. To investigate the various fundus findings in malignant arterial hypertension, I produced malignant arterial hypertension by modified Goldblatt’s procedure in one kidney in 60 adult rhesus monkeys; the procedure and various investigations are discussed at length elsewhere [6].
I could record only the systolic BP in these animals; the normal median systolic BP in the rhesus monkeys was 120 mmHg. I decided to consider that a systolic BP of 160 mmHg or greater was a hypertensive level. Renovascular hypertension developed in all 60 monkeys.
The following account of hypertensive fundus changes is essentially based on that experimental study, supplemented by my clinical experience of patients with malignant arterial hypertension.
The important conclusions on fundus changes in malignant hypertension which emerged from my study [6] were these:
1.
The ophthalmoscopic, fluorescein angiographic, and pathologic findings (see below) of this experimental study clearly showed that the various fundus changes seen in rhesus monkeys with malignant arterial hypertension fall into three distinct categories: (1) hypertensive retinopathy, (2) hypertensive choroidopathy, and (3) hypertensive optic neuropathy. I could find no information on the subject in the literature, particularly on the order of appearance of the three types of fundus changes and their relationship to the level of the BP.
2.
Investigation of the order of appearance of hypertensive retinopathy, choroidopathy, and optic neuropathy showed that the appearance of retinopathy was significantly (P < 0.01) earlier than that of choroidopathy or optic neuropathy, with no significant difference in the appearance time between the latter two (Fig. 23.4). However, there was no pattern to the order in which the three categories of fundus changes reached their maximum severity.
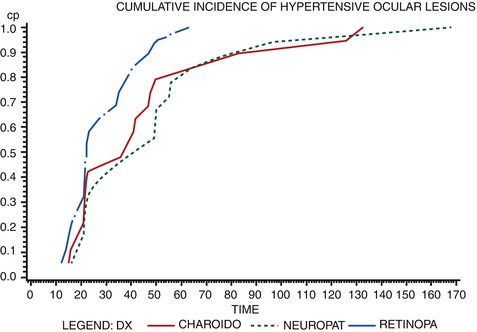
Fig. 23.4
A graphic representation of cumulative incidence of development of hypertensive retinopathy, choroidopathy, and optic neuropathy in relation to time from the renal artery clamping in experimental monkey model (Reproduced from Hayreh et al. [6])
3.
There was no statistically significant difference between the mean systolic BPs when hypertensive retinopathy, choroidopathy, and optic neuropathy first appeared and also when they were most marked. When the grades of severity of hypertensive retinopathy, choroidopathy, or optic neuropathy were correlated with the level of BP, no significant correlation was seen. These findings suggest that once the BP was elevated to the appropriate critical level, it resulted in the development of the various fundus lesions and the lesions progressed to their maximum severity while that level of BP was maintained. Thus, different levels of BPs were not required to produce different types of fundus lesions, and a higher BP was not required to produce more marked fundus lesions. This tends to conflict with the usual impression among clinicians that the higher the BP, the more marked the fundus changes.
4.
This study showed that when the rise in BP developed faster, the fundus lesions appeared significantly earlier (p = 0.00001). This would suggest that the ocular vascular bed may be able to adapt to a slow rise in BP without decompensating. The situation is probably similar to that of the corneal response to raised intraocular pressure; i.e., if the rise in the intraocular pressure is acute and sudden, corneal epithelial edema develops rapidly, but if the rise in the intraocular pressure is gradual, coming on over months, the cornea remains perfectly clear in spite of the much higher sustained intraocular pressure. The findings of this study suggest that the incidence of severity of the fundus lesions is also determined by factors other than the absolute levels of the BP. I could find no apparent relationship between the changes in sodium, chloride, potassium, blood urea nitrogen, or creatinine levels and the fundus lesions.
Among patients with malignant arterial hypertension and “hypertensive retinopathy,” renovascular malignant hypertension is a common finding [34, 35]. Thus, my findings on the fundus changes in rhesus monkeys with renovascular malignant arterial hypertension should hold good in man. It is well established now that renovascular hypertension in man and experimental animals is due to increased liberation of renin, which acts on angiotensinogen and liberates angiotensin I; the latter yields angiotensin II by the action of the converting enzyme. Angiotensin II is a powerful vasopressor substance and it also potentiates the vasoconstrictor activity of norepinephrine; the latter is released in excessive amounts due to angiotensin stimulating the sympathetic nervous system.
Hypertensive Choroidopathy
In the past, the primary objective of almost all clinical, pathologic, and experimental studies on ocular findings in malignant arterial hypertension has been to investigate changes in the retina and retinal vessels, with only a passing reference to other lesions. In 1855, von Graefe [36] first described serous retinal detachment in toxemia of pregnancy; since then choroidal lesions in malignant hypertension have been noted in a number of reports, mostly anecdotal in nature and usually in patients with toxemia of pregnancy or renal disease. Unfortunately, most of these ophthalmoscopic lesions were generally considered to be manifestations of retinopathy. In severe nephritis, Elschnig [37] has been credited with the discovery, in 1904, of the spots which bear his name; however, similar lesions associated with “albuminuria” were in fact described by much earlier authors, for example, Lecorche [38] in 1858 and Bousseau [39] in 1868. Clinical studies of these spots have shown them to be present almost invariably in conditions associated with malignant arterial hypertension; histopathological studies revealed these as areas of retinal pigment epithelial (RPE) proliferation in the center, surrounded by RPE atrophy, representing focal ischemic infarcts due to acute occlusion of the choriocapillaris or small choroidal arterioles [10, 11, 40–49]. “Siegrist’s streaks or spots” have been described as one of the fundus lesions of hypertension (although one of the two patients described by Siegrist with this lesion had giant cell arteritis and not arterial hypertension); [50] these represent chains of pigmented spots arranged like a string of beads lying on sclerosed choroidal vessels. In addition to the pathologic studies on Elschnig’s spots (mentioned above), there have been a number of other histopathological studies over the past 160 years describing hypertensive choroidal vascular changes associated with occlusion of the choroidal vessels and other lesions in patients with malignant arterial hypertension, with or without renal disease [38–63]. Based on pathologic studies, it has been suggested that choroidal vascular changes play an important role in the production of fundus changes in malignant hypertension and in visual disturbances [51, 53, 56]; Cohen [56, 57], and Harry and Ashton [59] stated that the severe forms of hypertensive retinopathy are accompanied by even more pronounced changes in the choroidal vessels. Thus, although the pathologists have been aware of the marked involvement of the choroidal vascular bed in malignant hypertension over the past 160 years, the clinicians have still been largely unaware of it, in spite of the fact that Duke-Elder [64] stated, more than 60 years ago, that, “In cases of advanced nephritis, always associated with albuminuria and usually with an extremely high BP, choroidal changes may become ophthalmoscopically visible to constitute a clinical picture of albuminuric choroiditis. These appearances are usually a terminal event in a protracted illness.” He classified the clinical lesions seen in “albuminuric choroiditis” into the following four groups: (1) pale yellow or reddish plaques, sometimes surrounded by pigmentary deposits, situated in the periphery of the fundus; (2) Elschnig’s spots [37]; (3) Siegrist’s streaks [50]; and (4) large patches of chorioretinal atrophy.
Since there was no detailed, systematic, longitudinal study on the clinical manifestations or on the histopathological lesions and clinicopathological correlation of hypertensive choroidal vascular lesions and their pathogeneses, I did a comprehensive investigation of this in my experimental study with renovascular malignant arterial hypertension in rhesus monkeys. The findings of this study are discussed at length elsewhere [10]. Following is a brief account. Since there is not much information available to ophthalmologists on the subject, I need to discuss in detail the types, nature, evolution, and pathogeneses of various lesions seen in hypertensive choroidopathy, as well as their ophthalmoscopic and fluorescein angiography findings.
In hypertensive choroidopathy, on ophthalmoscopy, serous retinal detachment (RD) and RPE lesions were seen. In addition to the two ophthalmoscopic findings, the eyes also showed choroidal vascular bed abnormalities, which were best seen on fluorescein angiography [10] and on histopathological findings [11].
Fluorescein Fundus Angiographic Abnormalities of the Choroidal Vascular Bed
In the early stages, the choroidal vascular bed showed mild to marked delayed or patchy filling, usually most marked in the central part of the macular region (Figs. 23.5 and 23.6). The choroidal filling abnormalities in the two eyes of a monkey were usually similar in severity. These abnormalities were seen in all monkeys, first detected when the BP was 180–190 (median) mmHg.
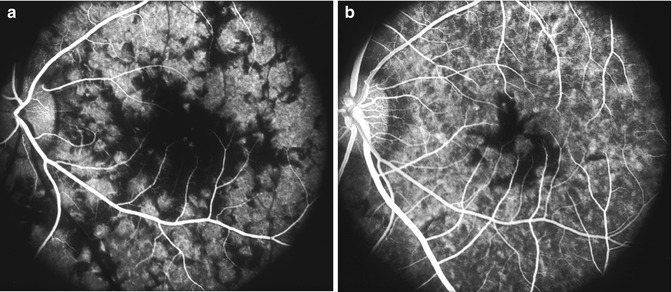
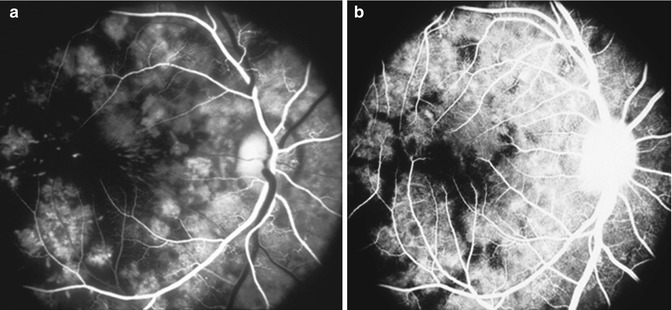
Fig. 23.5
Fluorescein fundus angiograms of the left eye in a monkey with BP 205 mmHg, during (a) retinal arterial and (b) late venous phases, show marked delay in filling of the submacular choroid (Reproduced from Hayreh et al. [10])
Fig. 23.6
Fluorescein fundus angiograms of the right eye in a monkey with BP 190 mmHg, during (a) retinal arterial and (b) postvenous phases, show marked delay in filling of the submacular choroid (Reproduced from Hayreh et al. [10])
After this, the filling defects frequently became more marked. In almost all of the monkeys that survived for a long time, the choroidal filling abnormalities persisted till the end of the follow-up period. There was a significant correlation between the time of onset of choroidal circulatory disturbances and when these were most marked (p < 0.01) and also between the BPs at the two stages (p < 0.01). Normally, it is not possible to see the choroidal vascular filling peripheral to the equator for some unknown reason [65], and thus I could not evaluate the choroidal circulation in that region; however, if the RPE is missing or degenerates in that area, then the choroidal vascular bed is unmasked.
Hypertensive Choroidopathy Lesions
Clinically, these consist of the following:
I.
Retinal pigment epithelial lesions (RPE): These are of two types
1.
Acute focal ischemic RPE lesions
2.
RPE degenerative lesions
II.
Serous retinal detachment (RD)
III.
Choroidal vascular bed abnormalities
Ischemic RPE lesions and serous RD may develop individually or collectively as the initial evidence of hypertensive choroidopathy.
Retinal Pigment Epithelial Lesions
These could be subdivided into (1) acute ischemic focal lesions and (2) degenerative lesions.
Acute Ischemic Focal RPE Lesions
These lesions were almost invariably punctate, round, focal, pale or white in color, mostly pinhead in size, and frequently distributed in clusters, although some varied in size (Figs. 23.7, 23.8, 23.9, and 23.10). The lesions were almost always situated in some part of the macular region (mostly in the central and/or temporal part) and occasionally nasal to the optic disc or elsewhere in the posterior fundus. The lesions also involved the peripheral part of the fundus (Fig. 23.10b); however, I could not document these lesions as frequently in the periphery as at the posterior pole in spite of the fact that chronic degenerative lesions were seen very commonly in the peripheral fundus in these monkeys (Figs. 23.11 and 23.12e, f); this was probably due to limitations in my ability to examine and photograph satisfactorily the peripheral part of the fundus. The severity of the lesions usually tended to be similar in the two eyes of a monkey (Fig. 23.10). Frequently, the acute and the resultant RPE degenerative spots were mixed because of the rapid transition of the former to the latter type. Also, it was often difficult to differentiate the acute lesions from the late degenerative lesions. Evaluation of the acute lesions was made more difficult by the not uncommonly faint and inconspicuous nature of the small pinpoint lesions, particularly in the frequent presence of overlying serous RD. The RPE lesions progressively increased in number with time. Some of the acute punctate lesions had focal mild RD overlying (Figs. 23.8 and 23.9).
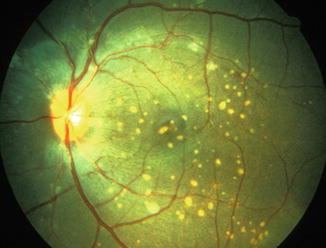
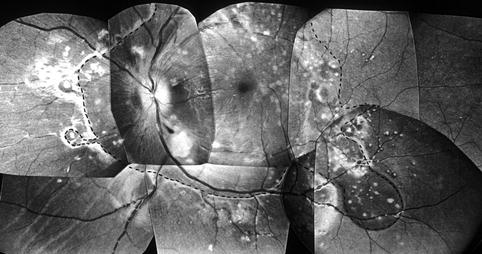
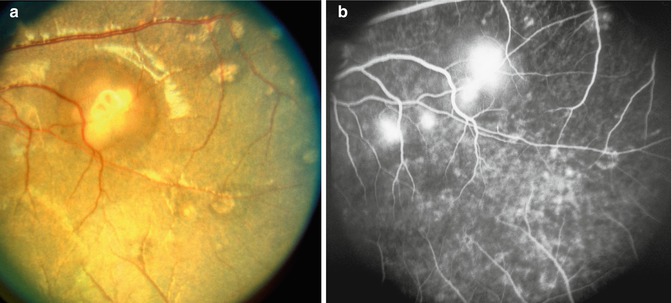
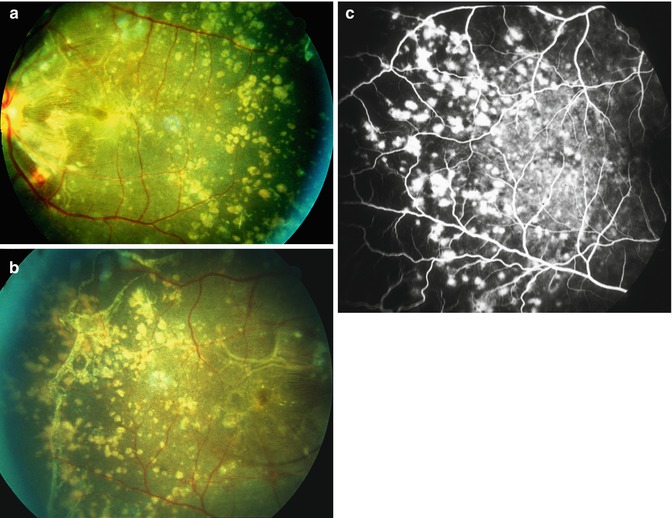
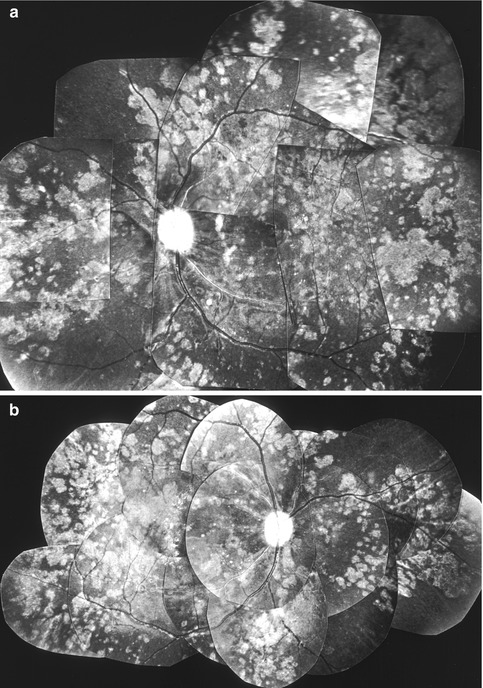
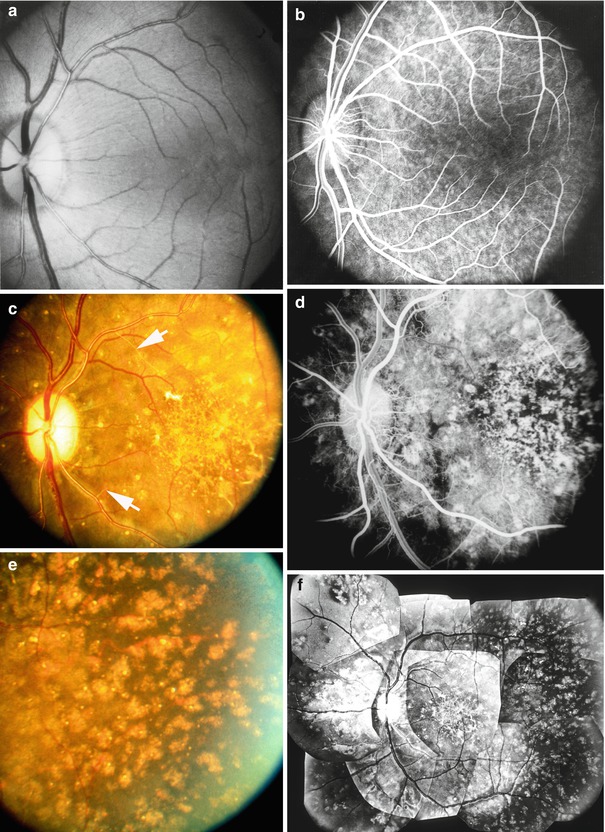
Fig. 23.7
Fundus photograph of the left eye of a monkey with BP 202 mmHg, shows acute focal RPE lesions, in addition to a few focal intraretinal periarteriolar transudates (FIPT) along the retinal arterioles (Reproduced from Hayreh et al. [10])
Fig. 23.8
Composite fundus photograph of the left eye of a monkey with BP 192 mmHg, shows multiple acute focal RPE lesions, extensive serous RD involving the entire macular and peripapillary region (as outlined by the dotted line), in addition to focal RD over some of the peripheral acute focal RPE lesions, two cotton-wool spots (above and below the optic disc), and occasional FIPT (Reproduced from Hayreh et al. [10])
Fig. 23.9
Fundus photograph (a) and fluorescein fundus angiogram (b), during the late phase, of the left eye of a monkey with BP 180 mmHg, showing multiple focal RPE lesions (acute lesions with overlying RD and many old lesions). On angiography, acute focal RPE lesions show fluorescein staining, whereas the old lesions show unmasking of choroidal fluorescence (Reproduced from Hayreh et al. [10])
Fig. 23.10
Fundus photographs of both eyes (a, left eye, b, right eye) and fluorescein angiogram (c) during the late phase of the right eye of a monkey with BP 190 mmHg, show multiple acute focal RPE lesions with serous RD and fluorescein staining of the acute RPE lesions (Reproduced from Hayreh et al. [10])
Fig. 23.12
Fundus photographs (a, c, e, f) and fluorescein fundus angiograms (b, d) of the left eye of a monkey. (a, b) Normal appearance before Goldblatt’s procedure. (c–f) with BP 220 mmHg with abnormal fundus and angiogram. (f) A composite fundus photograph. (c, f) Fundus photographs show retinal arteriolar sclerosis, increased arteriolar tortuosity and sheathing of fine arterioles (arrows in c), intraretinal microvascular abnormalities, retinal atrophy, and focal and macular RPE degenerative change; compare the state of the retinal vessels and of the fundus background in (c, f) with that in (a). (e) The temporal peripheral fundus with RPE degenerative change and drusen. (d) Angiogram during the retinal arteriovenous phase shows extensive retinal capillary obliteration, occlusion of the sheathed retinal arteriole, and widely scattered RPE degenerative change; note the angiographic changes as compared to the normal in (b) (Reproduced from Hayeh et al. [14])
Fluorescein fundus angiography at this stage almost always showed a variable degree of generalized delayed filling of the choroidal vascular bed, frequently patchy filling, with the macular or foveal region showing especially marked delay (Figs. 23.5 and 23.6) (the reason for that is discussed below). The acute lesions stained during the late phase (Figs. 23.9b and 23.10c), but the late degenerative lesions showed unmasking of the choroidal fluorescence during the transit of the dye (Figs. 23.13 and 23.14) and no staining during the late phase, whereas others showed no apparent angiographic abnormality. The acute punctate focal RPE lesions (on ophthalmoscopy) somewhat resembled focal intraretinal punctate transudates (FIPT discussed below – Fig. 23.15) [7], but on fluorescein angiography, the lesions stained less intensely and their staining persisted for a much shorter duration than the FIPTs’, apart from the fact that the FIPTs were intraretinal and the RPE lesions were subretinal.
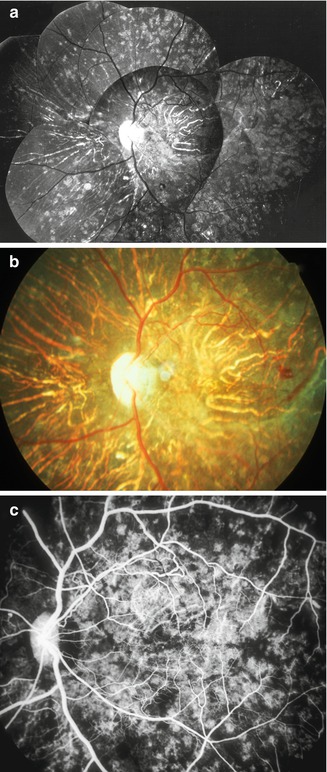
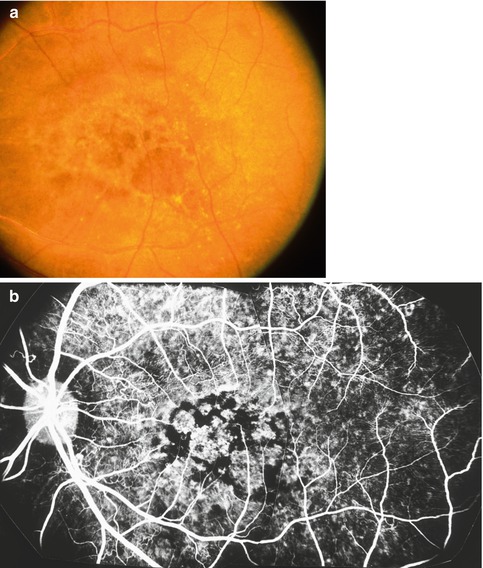
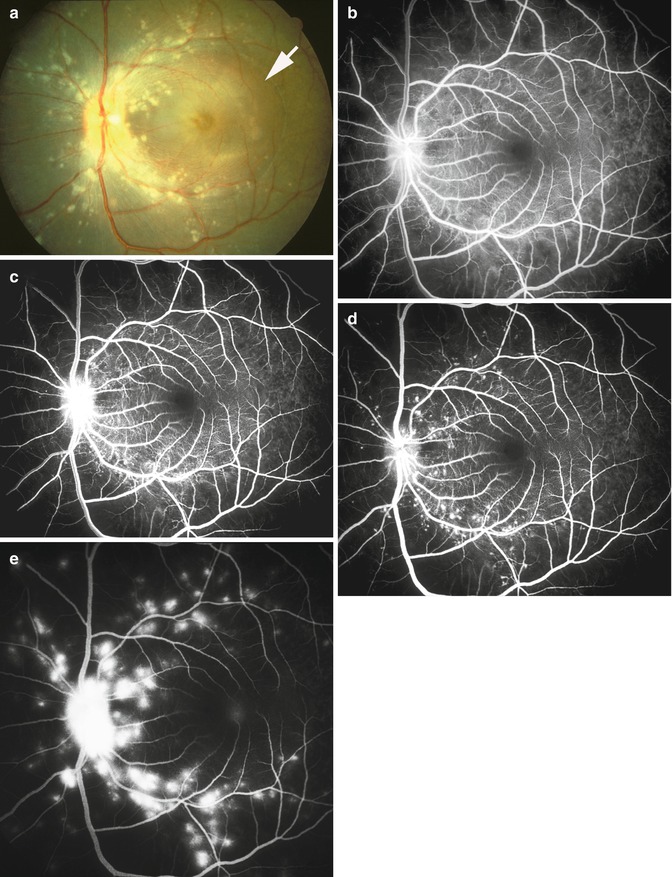
Fig. 23.13
(a) Composite and (b) color fundus photographs, and (c) fluorescein fundus angiogram (during retinal arteriovenous phase) of the left eye of a monkey with BP 195 mmHg, show extensive late RPE degenerative lesions with choroidal vascular sclerosis. Notice in the angiogram the filling of the sclerotic choroidal arteries, patchy filling of choroid, and unmasking of choroidal fluorescence at sites of RPE degeneration (Reproduced from Hayreh et al. [10])
Fig. 23.14
Fundus photograph (a) and fluorescein angiogram (b, during retinal arteriovenous phase) of the left eye of a monkey with BP 225 mmHg, show RPE degeneration in the macular region, resembling senile macular degeneration (Reproduced from Hayreh et al. [10])
Fig. 23.15
Fundus photograph (a) of the left eye of a monkey with BP 204 mmHg shows multiple dull white punctate retinal opacities (FIPTs) along major retinal arterioles and their main branches. It also shows macular retinal detachment (RD) – arrow outlines its boundary. Fluorescein fundus angiogram (b–e): (b) 1.15 s, (c) 12.5 s, (d) 15.5 s, and (e) 155 s after the injection of the dye. Note evolution of fluorescein filling and leaking spots corresponding to punctate retinal opacities of FIPTs in (a) (Reproduced from Hayreh et al. [7])
RPE Degenerative Lesions
The lesions could be further subdivided into early and late types.
(a)
Early RPE Degenerative Lesions: In 2–3 weeks, the acute focal RPE lesions evolved into focal RPE degenerative lesions. Thus, like the acute lesions, the initial degenerative lesions were most commonly seen in some part of the macular region and much less frequently elsewhere in the posterior and/or peripheral fundus. When the acute lesions were focal in nature, they were replaced by similar-looking depigmented spots. Often, the ophthalmoscopic appearance of the acute and early degenerative lesions was so similar that it was almost impossible to differentiate the two, but fluorescein angiography showed unmasking of the underlying choroidal fluorescence with no late staining of the degenerative lesions. The confluent acute lesions changed into either ill-defined chorioretinal degenerative areas or fairly defined, punched-out RPE degenerative lesions.
(b)
Late RPE Degenerative Lesions: The degenerative RPE lesions were almost invariably progressive in nature. The progression of the lesions was much more rapid and marked during the early stages. These findings strongly indicated that the progressive degenerative lesions were due to persistent, chronic choroidal ischemia, not always preceded by the acute lesions. This could be the reason why the chronic degenerative lesions were almost invariably far more extensive than the acute lesions. The late degenerative lesions were usually widely scattered throughout the fundus, distributed maximally in the macular and peripheral parts, and much less frequently in other parts (Figs. 23.11, 23.12, 23.13, and 23.14). In the macular region, the lesions were most marked in the temporal part and not infrequently also in the foveal area. The peripheral fundus was usually involved much more extensively than the macular area, and the temporal periphery was usually maximally involved. Thus, the temporal part of the macular region and the temporal part of the peripheral fundus were most involved by these lesions, whereas the rest of the fundus was much less involved.
The degenerative lesions were usually composed of polymorphic RPE atrophic lesions as well as of diffuse pigmentary change, the latter giving a coarse, granular, or moth-eaten appearance to the RPE (Figs. 23.11, 23.12, 23.13, and 23.14). The atrophic lesions varied from being focal in nature to confluent patches of varying shapes (e.g., geographic, triangular, irregular, or other) and sizes (as the time passed some of the atrophic lesions tended to become confluent). Some of the focal depigmented lesions were surrounded by a dark halo (Figs. 23.9a and 23.12e, f), and at other places, depigmented patches were lying adjacent to hyperpigmented patches, and occasionally big gray patches developed; similar lesions were seen after occlusion of the posterior ciliary arteries (see Chap. 18). Elschnig’s spots were seen. Siegrist’s streaks rarely developed. Some eyes showed drusen, which, in a few eyes, had a white glistening appearance (Fig. 23.12e, f). In the macular region, the changes were typically like those usually seen in age-related macular degeneration, with moth-eaten looking RPE (Figs. 23.11, 23.12, 23.13, and 23.14), and less frequently subretinal fibrosis (from hyperplastic metaplasia of the RPE – Fig. 23.16), subretinal gliosis, drusen, and other RPE degenerative changes. In some of the eyes, the fundus picture very much resembled the late stages of the clinical entity called “bird shot retinopathy.”
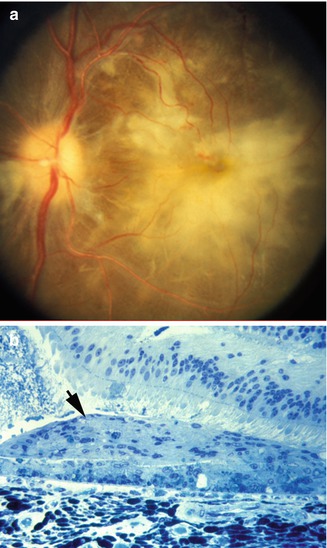
Fig. 23.16
Fundus photograph (a) of the left eye of a monkey with BP 250 mmHg, shows subretinal fibrosis in the macular region; this eye previously had serous RD. (b) Histological section showing subretinal fibrosis (arrow) from RPE metaplasia (Reproduced from Hayreh et al. [10])
Fluorescein fundus angiography showed the RPE degeneration much more clearly than ophthalmoscopy because of unmasking of the underlying choroidal fluorescence in the degenerated areas (Figs. 23.13c, and 23.14b). The angiography, therefore, usually revealed much more extensive lesions than were seen on ophthalmoscopy, although occasionally an ophthalmoscopically visible lesion showed no angiographic abnormality. There was no late staining of the lesions apart from the staining of the drusen. Hyperpigmented areas and some of the whitish lesions showed masking of the choroidal fluorescence (Fig. 23.14). In eyes where the macular region was involved, the angiographic appearance was very much like age-related macular degeneration.
The RPE lesions seen in this study were similar to those in my study in experimental posterior ciliary artery occlusion [66].
Serous Retinal Detachment
Among the monkeys that had hypertensive choroidopathy, there was a serous RD of variable extent and severity. It almost invariably involved the retina in the macular and/or peripapillary region and occasionally the peripheral retina.
Macular and/or Peripapillary Serous RD
RD was most common in the macular region, often involving the entire macular region (Figs. 23.8, 23.15, 23.17, 23.18, 23.19, 23.20, and 23.21) but occasionally located only in the foveal zone. RD in the peripapillary region was a common finding and it occurred alone or in combination with the macular RD. The extent of the RD in the two eyes was usually similar in bilateral cases. In addition to the well-developed macular serous RD, small localized blister-like spots of serous RD were also frequently seen over the acute ischemic focal RPE lesions in the posterior fundus (Figs. 23.8 and 23.9a).
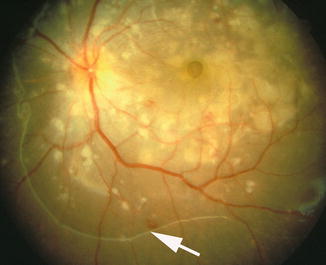
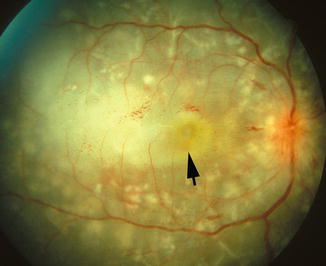
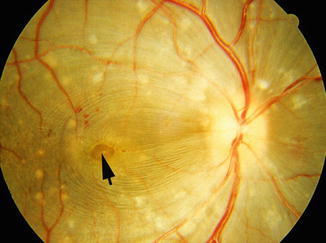
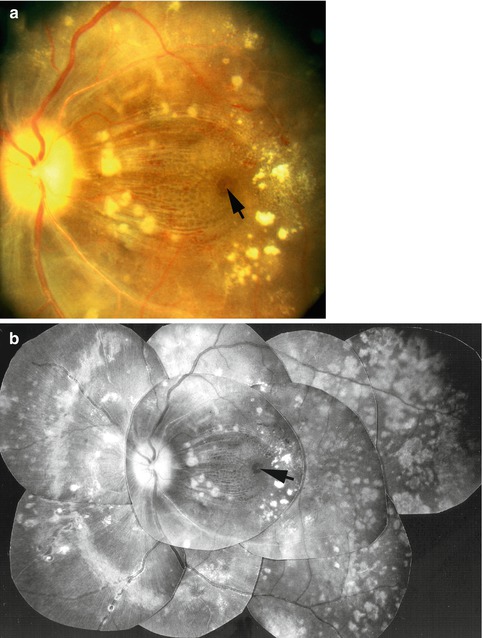
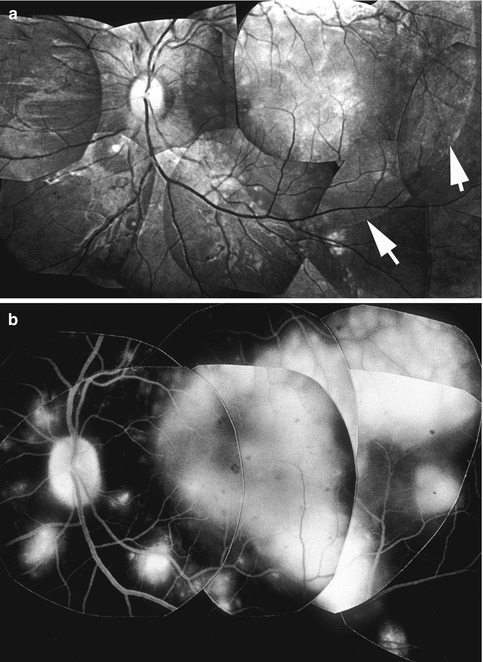
Fig. 23.17
Fundus photograph of the left eye of a monkey with BP 198 mmHg shows serous RD of posterior pole (notice arrow outlining the RD) with multiple acute focal RPE lesions, foveal cyst, and multiple FIPTs (along the retinal arterioles) (Reproduced from Hayreh et al. [10])
Fig. 23.18
Fundus photograph of the right eye of a monkey with BP 198 mmHg shows multiple acute focal RPE lesions, serous RD of macular region with proteinous opaque subretinal fluid (arrow), and multiple FIPTs (along the retinal arterioles)
Fig. 23.19
Fundus photographs of the right eye of a monkey with BP 190 mmHg shows serous RD of macular region associated with macular edema, separation of nerve fibers by edema, microcystic edema, and foveal cyst (arrow). Other lesions of hypertensive choroidopathy and retinopathy are also seen (Reproduced from Hayreh et al. [10])
Fig. 23.20
Fundus photographs (a color and b composite) of the left eye of a monkey with BP 212 mmHg show serous RD of macular region associated with macular edema, separation of nerve fibers by edema, microcystic edema, foveal cyst (arrow), cotton-wool spots, lipid deposits, extensive late RPE degenerative lesions, optic atrophy, and retinal arteriosclerosis (Reproduced from Hayreh et al. [10])
Fig. 23.21
Composite fundus photograph (a) and fluorescein fundus angiogram (b, during the late phase) of the left eye of a monkey with BP 206 mmHg shows serous RD in the macular region (arrows) with acute focal RPE lesions, and a few FIPTs (along the retinal arterioles). In (b) notice fluorescein staining of the subretinal fluid, acute RPE lesions, and FIPTs (Reproduced from Hayreh et al. [10])
When first recorded, the RD in the macular region was flat and mild. The peripapillary RD was always flat and mild. The time of development of the RD was significantly (P = 0.01) later than the onset of angiographically seen choroidal circulatory disturbances. The acute ischemic RPE lesions developed earlier than the onset of the serous RD. There was a significant correlation (P < 0.01) between the BPs when the three types of changes (i.e., choroidal circulatory disturbances, acute RPE lesions, and RD) were first seen. When macular and/or peripapillary serous RD was first seen, all of the eyes with the RD had already had choroidal circulatory disturbances for a variable length of time. Findings showed no definite relationship between serous RD and hypertensive retinopathy.
The macular RD usually increased in severity during the initial stages. When the RD was marked, it involved either the entire posterior pole (including the peripapillary region) or most of the macular region. It was usually bullous in nature, with the central part (almost invariably in the foveal region) often bulging like a nipple. When the RD was marked, the retina in that area frequently looked diaphanous. Occasionally, outpouching of the RD at its peripheral margin was seen over the acute focal RPE lesions (Fig. 23.8). At the last examination, which was at a variable time after the onset of the RD as well as the hypertension, the RD in the macular region had resolved completely in about half of the monkeys (e.g., compare Figs. 23.11, 23.12, and 23.13 with Figs. 23.8, 23.17, 23.18, 23.19, 23.20, 23.21, and 23.22).
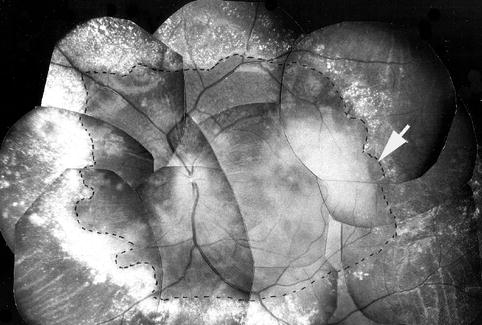
Fig. 23.22
Fundus photograph of the left eye of a monkey with BP 220 mmHg shows serous RD of the posterior pole (outlined by dotted line – arrow) with extensive white deposits peripheral to the RD. Whitish material in the macular region is subretinal proteinous deposit on the RPE (Reproduced from Hayreh et al. [10])
The macular retina mostly showed frank edematous changes. Frequently with microcystic changes, a prominent foveolar cyst developed in a quarter of the monkeys (Figs. 23.17, 23.19, and 23.20). As a manifestation of the retinal edema over the RD, some of the eyes showed separation of the nerve fibers, mainly in the region of the papillomacular bundle (Figs. 23.19 and 23.20). In a number of the eyes, the retina over a detached area was diaphanous or atrophic. Occasionally, when the RD resolved, retinal folds developed. As an example, evolution of macular retinal changes in one eye is shown in Figs. 23.8, 23.10a, 23.20, and 23.11a, on days 63, 78, 174, and 245, respectively.
The subretinal fluid in all the monkeys at the onset of RD was almost always clear (Figs. 23.17 and 23.19), with a variable amount of turbidity in the foveal zone in the majority. The fluid tended to become progressively more turbid in nature with time and was thick, gelatinous or dense, opaque, white, and exudative in nature in marked cases, most marked in the foveal zone (Figs. 23.18 and 23.22). In some of the eyes, the exudative material condensed to form a membranous deposit over the RPE (Fig. 23.22). The subretinal fluid or opaque material usually did not block the underlying choroidal fluorescence on fluorescein angiography, but in about a quarter, it stained with fluorescein because of the leakage of the dye from the underlying choroid (Fig. 23.21b). The exudative material started to clear slowly, almost always in a centripetal fashion from the periphery. It was replaced by clear fluid, with the central foveal zone showing a variable amount of subretinal opacity in most cases, even at termination. In a number of eyes with thick proteinous-looking exudate in the subretinal space, this material resolved in patches or in a moth-eaten pattern so that it gave the macular region a honeycomb cystoid appearance on ophthalmoscopy. One third of the eyes showed a subretinal deposition of white opaque tissue over the macular RPE (Fig. 23.16); in most of these eyes, the RD had already resolved completely. The type and extent of subretinal fluid and/or deposit showed no apparent correlation with the retinopathy, the choroidal angiographic findings, or the RPE lesions. The early and late changes in the RPE showed a good correlation with the onset, development, and severity of macular serous RD.
Peripheral Serous RD
An additional peripheral serous RD developed in about a third of the eyes, after a significantly longer time than that of the onset of acute focal RPE lesions (p = 0.04). The peripheral RD when present involved 360° of the peripheral retina and was occasionally not connected to the posterior polar RD. The RD was almost invariably bullous in nature, and in total RD, the subretinal fluid shifted markedly with the position of the head, and the retina usually showed folds (Fig. 23.23). The subretinal fluid was usually clear, but in some eyes it was turbid and in a few even had subretinal whitish narrow fibrin bands (Fig. 23.10b). At the final examination, peripheral RD had completely resolved in the majority. All the eyes with peripheral RD usually showed marked choroidal circulatory disturbance. Once the RD resolved, the eyes almost always showed extensive RPE degeneration.
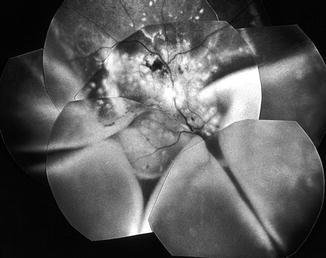
Fig. 23.23
Fundus photograph of the right eye of a monkey with BP 212 mmHg shows multiple acute focal RPE lesions and massive bullous serous RD involving the posterior pole and lower part of the peripheral retina, with retinal folds (Reproduced from Hayreh et al. [10])
Choroidal Vascular Bed Abnormalities
Normally, choroidal vessels are not seen on ophthalmoscopy. In some of the eyes with extensive RPE degenerative lesions, the large choroidal vessels became unmasked and were seen as white lines due to choroidal sclerosis (Figs. 23.13a, b); on fluorescein angiography they showed blood flowing in them (Fig. 23.13c). Fluorescein fundus angiography invariably revealed disturbances in the choroidal blood flow, as discussed above in the angiographic section (Figs. 23.5 and 23.6).
Pathologic Study
The pathologic features of hypertensive choroidopathy in our experimental monkeys may be categorized into three phases: (1) acute ischemic, (2) chronic occlusive, and (3) chronic reparative, depending on the duration and severity of hypertension. These are described in detail elsewhere [11]. Following is a brief account.
Acute Ischemic Phase: Pathologically, the retinal arterioles appeared unaltered. In contrast, choroidal arterioles were constricted and obliterated. The choriocapillaris adjacent to occluded arterioles exhibited necrotic endothelial cells that desquamated into the lumen; however, the lumens of the choriocapillaries remained patent. Fibrinous deposits were seen at Bruch’s membrane. In addition, the overlying RPE adjacent to occluded arterioles was vacuolated and subretinal exudate was seen. The RPE demonstrated intercellular edema, vacuolated endoplasmic reticulum, and loss of basal infoldings and apical microvilli.
Chronic Occlusive Phase: Pathologically, choroidal arteries and arterioles showed marked hyperplastic changes. The lumina of the choroidal arteries were extremely narrowed (Fig. 23.24a) or occluded (Fig. 23.24b). Proliferated intimal cells surrounded by an internal elastic membrane looked like an onion skin (Fig. 23.24c) and occluded the vessel lumen (Fig. 23.24d). Occluded choroidal arterioles were common. The choriocapillaries were diffusely occluded by fibrin thrombi. The overlying RPE was necrotic. The HRP study revealed staining in the degenerated RPE.
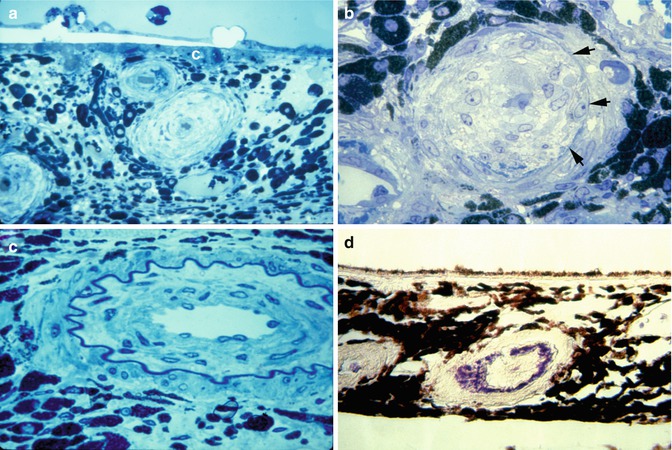
Fig. 23.24
Choroidal arteries in chronic occlusive phase of hypertensive choroidopathy. (a) Choroidal arteries and arterioles show marked hyperplastic arteriosclerosis. Lumens are extremely narrowed by thickened and laminated vessel wall. Choriocapillaris (c) is occluded by thrombus (toluidine blue, original magnification ×200). (b) Intimal cells proliferated within internal elastic lamina (arrows) have occluded lumen. Media (m) are attenuated and stretched around thickened intima (toluidine blue, original magnification ×420). (c) Proliferated intimal cells surrounded by internal elastic lamella show onion skin appearance (toluidine blue, original magnification ×520). (d) Fibrin deposit is seen in choroidal arterial wall (phosphotungstic acid-hematoxylin, original magnification ×280) (Reproduced from Kishi et al. [11])
Chronic Reparative Phase: The occluded choroidal arteries were recanalized. At the level of the arterioles, recanalization was also seen. Patent choriocapillaries became sparse. In this phase, regenerated RPE was hypopigmented and attenuated and showed extensive endoplasmic reticulum. At the border of the retinal detachment, RPE demonstrated placoid proliferation [60].
In summary, our pathologic study [11] showed that focal necrosis of the RPE (Elschnig’s spots) developed under a patent choriocapillaris but constricted arterioles. We believe that the pathogenesis of Elschnig’s spots may be related to the lobular arrangement of the choriocapillaris, whereby each is fed centrally by small arterioles and drained peripherally by veins [67] (Fig. 23.25, see Chap. 4). The fact that severe constriction of central arterioles produced focal necrosis of the RPE without involving the adjacent choriocapillaris supports the concept that each lobule of the choriocapillaris functions as an independent circulatory unit. Pathologic study demonstrated that occlusive changes involving choroidal arteries (Fig. 23.24), arterioles, and the choriocapillaris were responsible for the extensive necrosis of the RPE that resulted in RD. Our study showed leakage through the RPE into the subretinal space. The primary change in choroidal arteries and arterioles is fibrinoid necrosis (Fig. 23.24d). The choroidal vasculature responds to systemic hypertension differently from the retinal circulation, because the latter is autoregulated and the former is controlled by sympathetic nerve tone. The changes in the choroidal circulation also play an important role in the production of hypertensive optic neuropathy, as discussed below.
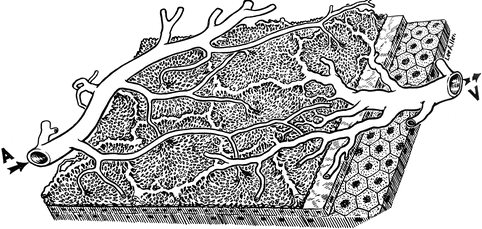
Fig. 23.25
A three-dimensional schematic representation of the choriocapillaris pattern. A choroidal arteriole, V choroidal veins (Reproduced from Hayreh [67])
Our experimental studies on malignant arterial hypertension have clearly shown that hypertensive choroidopathy is an important and distinct clinical entity.
Pathogenesis of Hypertensive Choroidopathy
From all the available evidence from my experimental studies (clinical and pathologic [11]) and from previously reported clinical and pathologic studies in patients with malignant arterial hypertension, it can be concluded that hypertensive choroidopathy is primarily due to choroidal ischemia, which produces ischemic damage of the overlying RPE, resulting in the development of the various ophthalmoscopically visible lesions.
Mechanism of Choroidal Ischemia
As discussed above, the renin-angiotensin-aldosterone system plays an important role in the development and maintenance of experimental renovascular arterial hypertension. Angiotensin II is a powerful vasopressor substance and it also potentiates the vasoconstrictor activity of norepinephrine, which is released in excessive amounts due to angiotensin stimulating the sympathetic nervous system. It is well established that choriocapillaris possesses no blood-ocular barrier because its endothelium shows the presence of numerous fenestrations, which make the choriocapillaris freely permeable. Thus, in malignant hypertension, there is a marked leak of angiotensin and other materials from the choriocapillaris into the choroidal fluid, where angiotensin and norepinephrine act on the wall of the choroidal vessels and result in the choroidal vasoconstriction and ischemia. Sympathetic innervation of the choroidal arterioles may further make them more susceptible to vasoconstriction [68]. The choroidal ischemia is not an “all-or-none” phenomenon because it may vary from subclinical to extreme degrees. The fluorescein angiographic and pathologic studies described above showed that the choroidal ischemia initially is of an acute type and later becomes chronic; the effects of the two types of ischemia on the overlying RPE vary.
Acute severe choroidal ischemia causes focal infarction of the RPE and of the overlying outer retina, resulting in the production of acute focal RPE lesions (Figs. 23.7, 23.8, 23.9, 23.10, 23.17, 23.18, 23.19, and 23.21). Normally, RPE exercises a blood-retinal barrier, preventing leakage of fluid and other materials from the choroid to the retina. Infarction of the RPE causes breakdown of this blood-retinal barrier, resulting in leakage of fluid and other materials from the choroid under the retina. Fluorescein angiography revealed leakage of the dye from the choroid into the subretinal fluid (Figs. 23.9b and 23.21b). Horseradish peroxidase studies in these monkeys showed leakage of the tracer material from the choriocapillaris into the subretinal fluid (Fig. 23.26). These findings confirm the breakdown of the blood-retinal barrier in the RPE. Submacular choroid was the region most commonly and most markedly affected by choroidal ischemia in malignant hypertension in this study (Figs. 23.5 and 23.6). Serous RD of the macular region was a very frequent finding (Figs. 23.8, 23.10, 23.15, 23.17, 23.18, 23.19, 23.21, 23.22, and 23.23). The reason for the marked vulnerability of the macular region to choroidal ischemia in hypertensive choroidopathy is that watershed zones between multiple short posterior ciliary arteries meet in the submacular choroid (see Figs. 4.18 and 4.19 in Chap. 4). The role and mechanism of watershed zones, which are most vulnerable to vascular insufficiency under such circumstances, is discussed in Chap. 4. This is very well demonstrated by fluorescein angiographic study in hypertensive choroidopathy (Fig. 23.6).
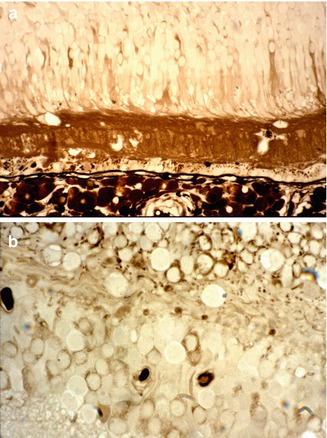
Fig. 23.26
Histological sections of an eye with serous RD and macular edema, after horseradish peroxidase (HRP) intravenous injection. Notice the leakage of HRP from the choriocapillaris into the subretinal fluid (in a) and into the retinal tissue (in a, b) (Reproduced from Hayreh et al. [10])
Development of serous RD is a well-known complication in toxemia of pregnancy, with many reports since the first description by von Graefe in 1855 [36]. The pathogenesis of RD in these cases has been controversial. Over the recent years, there have been anecdotal fluorescein fundus angiographic reports, of about a dozen patients in all [69–74]. During the acute phase, these eyes showed choroidal hypoperfusion and leakage of the dye into the subretinal fluid and, eventually, RPE degenerative change. Because patients with toxemia of pregnancy have associated malignant arterial hypertension, the available evidence indicates that the mechanism of RD in toxemia of pregnancy is similar to that seen in my study (i.e., choroidal ischemia, secondary to accelerated arterial hypertension, is the primary cause).
Acute multifocal choroidal ischemia is also seen in a number of other clinical conditions, apart from malignant arterial hypertension (e.g., disseminated intravascular coagulopathy, collagen vascular diseases, thrombotic thrombocytopenic purpura, leukemia, Goodpasture’s syndrome, hemolytic diseases, cardiac lesions, giant cell arteritis, and a host of other local vascular and systemic diseases); the subject was reviewed in detail by me elsewhere [75, 76]. Irrespective of the cause of choroidal ischemia, the RPE and other tissues supplied by the choroidal circulation can respond only in a limited fashion to the ischemia. Therefore, hypertensive choroidopathy can be confused with acute multifocal choroidal ischemic lesions from other causes and with various types of punctate epitheliopathies or choroidopathies.
In my study, during the late stages of evolution of the choroidopathy with advanced RPE degenerative lesions, the macular region in a number of the eyes showed changes indistinguishable from those seen in age-related maculopathy (Figs. 23.12c, d, 23.13, and 23.14), and drusen developed in some eyes (Fig. 23.12e, f). Drusen, in these eyes, were not an incidental finding but related to hypertensive choroidopathy and ischemia. I feel these findings may be relevant in explaining the pathogenesis of these lesions commonly seen in patients, indicating that chronic choroidal ischemia may play an important role in their development. This was further supported by recent findings of evidence of impaired blood supply in the macular regions in these eyes [77, 78]. As discussed above, the marked vulnerability of the macular region to choroidal ischemia in hypertensive choroidopathy and other conditions is because watershed zones between multiple short posterior ciliary arteries meet in the submacular choroid [78] (see Chap. 4).
Hypertensive Choroidopathy and Retinopathy
These are two independent and unrelated manifestations of malignant arterial hypertension. Usually the various manifestations of hypertensive choroidopathy have been considered to be part of retinopathy, and choroidopathy and retinopathy have been thought of as interrelated phenomena. We now know that this is a mistaken impression. As discussed above, the retinal and choroidal vascular beds have some fundamentally different properties, so that they respond very differently to malignant arterial hypertension, and consequently the hypertensive choroidopathy and retinopathy are two different and entirely independent phenomena.
Conclusion
This study shows that hypertensive choroidopathy is a distinct clinical entity. It is due to choroidal ischemia, with secondary ischemic changes in overlying RPE, which result in various RPE lesions and RD.
Hypertensive Optic Neuropathy
Although the clinical importance of optic disc edema in malignant arterial hypertension is well known, there is little definite information available on its clinical course, relationship to other hypertensive fundus changes and arterial BP in these patients, and, above all, on its pathogenesis. I did a comprehensive investigation of this in my experimental study with renovascular malignant arterial hypertension in rhesus monkeys. The findings of this study are discussed at length elsewhere [8, 9]. Following is a brief account.
My study [8] showed that hypertensive optic neuropathy is a distinct clinical entity, and provided useful information on the subject, as is evident from the following.
Optic Disc Changes
Optic Disc Edema
This was the initial evidence of hypertensive optic neuropathy. The disc, when edematous, showed not only swelling (Figs. 23.27, 23.28, 23.29a, 23.31, and 23.32) but also either hyperemia (Figs. 23.27 and 23.29a) or, sometimes, some degree of pallor (Figs. 23.28, 23.29b, 23.30a, 23.31, and 23.32), and on fluorescein angiography, there was staining of the disc during the late phase (Fig. 23.33) except in mild cases where the disc showed no definite staining. In this experimental study, the median BP when hypertensive optic neuropathy first appeared was 190 (197 ± 20 SD) mmHg. The time of initial appearance of the optic disc edema was significantly different (p = 0.0002) from that when it reached its maximum severity; the median BP at this stage was 196 (196 ± 26 SD) mmHg, and this was not significantly different from the BP when the optic disc edema first appeared (p = 0.9).
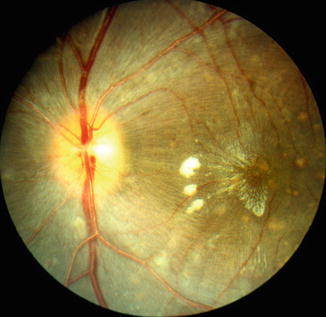
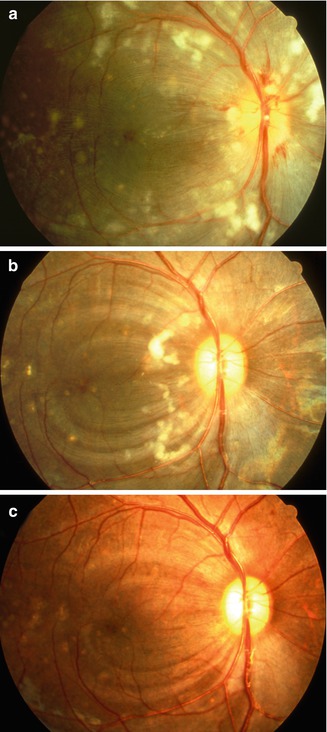
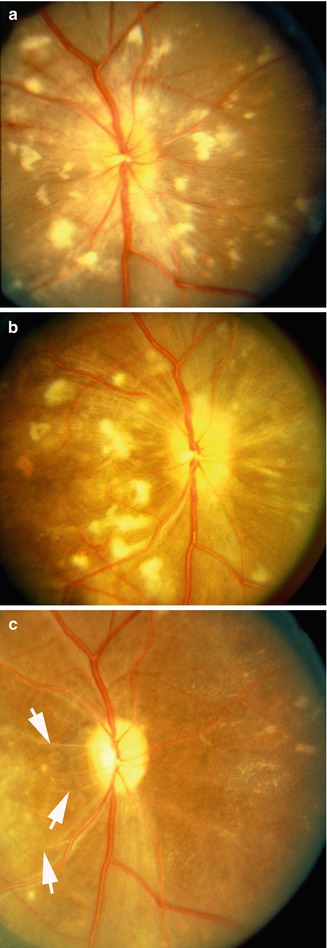
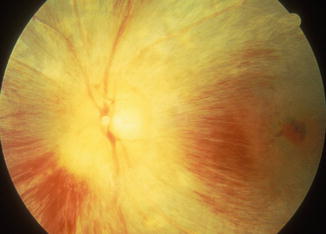
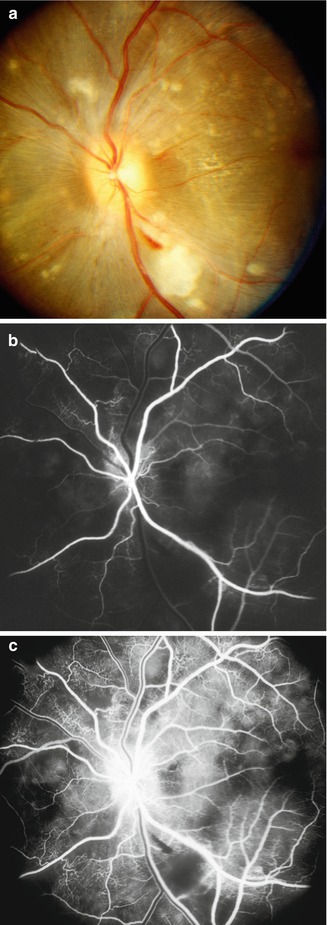
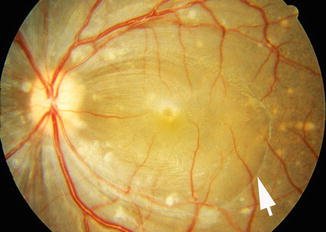
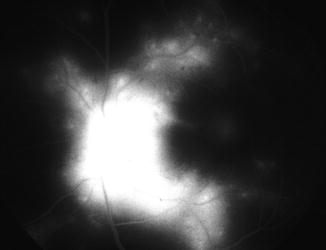
Fig. 23.27
Color fundus photograph of left eye of a monkey with BP 214 mmHg shows optic disc edema, focal RPE lesions, cotton-wool spots, and lipid deposits like a Japanese fan (Reproduced from Hayreh et al. [8])
Fig. 23.28
Fundus photographs of right eye of a monkey: (a) photograph with BP 190 mmHg shows multiple CWSs, FIPTs, acute focal RPE lesions, peripapillary retinal hemorrhages, and macular edema. (b) Shows that the old CWSs have resolved and new ones have appeared, with development of multiple areas of nerve fiber loss (seen as dark semicircular bands in the macular region). (c) Shows all CWSs have resolved, leaving multiple areas of nerve fiber bundle loss. Note also the presence of multiple focal RPE lesions in the temporal part of the macular region (a Reproduced from Hayreh et al. [8]; b, c reproduced from Hayreh et al. [15])
Fig. 23.29
Color fundus photographs of the right eye of a monkey 83 (a), 127 (b), and 293 (c) days after renal artery clamping with blood pressures of 190, 190, and 204 mmHg, respectively. Optic disc shows edema and hyperemia in (a), edema and pallor in (b), and atrophy in (c). Note also the presence of CWS, FIPTs, and arteriosclerotic change. In (c) arrows indicate the sheathed arterioles and in nasal part of the fundus a few lipid deposits and focal RPE lesions (Reproduced from Hayreh et al. [8])
Fig. 23.30
Fundus photograph of the left eye of a monkey with blood pressure of 190 mmHg shows marked optic disc edema and pallor, and retinal hemorrhages (Reproduced from Hayreh et al. [8])
Fig. 23.31
The left eye of a monkey with BP 192 mmHg. (a) Fundus photograph shows moderate optic disc edema and pallor, a small peripapillary hemorrhage, cotton-wool spots, FIPTs, and focal RPE lesions. (b, c) Fluorescein fundus angiograms: (b) during early retinal arteriovenous phase shows practically no filling of the choroid, and (c) during the arteriovenous phase shows no filling of the retinal capillaries in the distribution of the cotton-wool spots and optic disc staining (a, b Reproduced from Hayreh et al. [8]; c from Hayreh et al. [14])
Fig. 23.32
Left fundus photograph of a monkey with BP 190 mmHg shows moderate optic disc edema and pallor, FIPTs along retinal arterioles and on the optic disc, macular serous RD (arrow), and focal RPE lesions (Reproduced from Hayreh et al. [8])
Fig. 23.33
Fluorescein fundus angiogram of the left eye during late phase showing marked fluorescein leak in the optic disc and FIPTs
Superficial Hemorrhages on the Optic Disc or At/Near Its Margins
These were seen in only some of the eyes with optic disc edema (Figs. 23.28 and 23.31a); when present they were small and mostly seen when the disc was markedly edematous. Two eyes developed a marked venous stasis retinopathy secondary to marked optic disc edema and the disc hemorrhages were a part of that.
Correlation Between Initial Optic Disc Edema and Peripapillary Serous Retinal Detachment
Macular and peripapillary serous retinal detachment was a common finding in these eyes. This study indicated that the peripapillary retinal detachment and optic disc edema were two independent manifestations of malignant hypertension.
Pallor of the Optic Disc
The pallor initially was usually progressive (Fig. 23.29). The pallor appeared 77 (median) days after the onset of optic disc edema and 42 (median) days after the edema was most marked. There was no significant correlation between the severity of optic disc edema and that of optic atrophy, and this shows that the severity of optic disc edema was a poor guide to the severity of optic atrophy seen later on. I have also seen that in the human non-arteritic anterior ischemic optic neuropathy [79].
Fluorescein Fundus Angiographic Findings
Three-quarters of the eyes showed variable amounts of choroidal filling delay or defect in the posterior pole (Fig. 23.31b). Pathologic studies clearly demonstrated the universal presence of occlusion of the choroidal arteries, arterioles, and choriocapillaris as well as acute and chronic choroidal ischemic lesions (see above “hypertensive choroidopathy” [10]).
Histopathological Studies
To elucidate the pathogenesis of hypertensive optic neuropathy, we did histopathological study by light and electron microscopy with intravenous injection of horseradish peroxidase; it is discussed at length elsewhere [9]. The pathologic process may be conveniently categorized into three phases: acute ischemic, reparative, and atrophic. In the acute ischemic phase, the blood vessels in the surface nerve fiber layer of the optic nerve head showed constriction with intact endothelial cells but mildly degenerate pericytes. The pial vessels that supply the retrolaminar optic nerve, however, exhibited severe constriction with swelling of endothelial cells, and degeneration of pericytes and obliterated lumen were noted. The pial arterial system appeared to react more severely than the retinal vessels. In the resolution phase, the blood vessels in the surface nerve fiber layer showed no leakage of the tracer while the pial vessels showed extensive leakage into the connective tissue. In the atrophic phase, the differences between the vascular response in the surface nerve fiber layer and the retrolaminar myelinated nerve were even more dramatic. Ischemia appeared to be more severe in the myelinated optic nerve supplied by the pial arterial system than in the surface nerve fiber layer supplied by the central retinal artery.
In this experiment, hydropic swelling of axons without accumulation of organelles but with disruption of axolemma in the surface nerve fiber layer and prelaminar regions appears to be a consequence of ischemic infarct. Glial swelling is further supportive evidence. These morphologic features differentiate ischemic hypertensive optic neuropathy from optic disc edema in raised intracranial pressure [80, 81].
Ischemia was prominent in the optic nerve head in these eyes. This suggests that ischemia was multifocal, sparing some axons anteriorly but infarcting more posteriorly by the severe occlusion of the arterioles of the myelinated nerve. In addition, anatomic narrowing [82, 83] of axonal bundles in the lamina cribrosa might have further retarded the axoplasmic transport. It appears that the optic disc swelling in severe hypertension is essentially due to ischemic hydropic swelling of axons. Extensive gliosis was observed in the prelaminar optic nerve in the atrophic phase. In contrast, glial degeneration was seen without extensive glial proliferation in the retrolaminar optic nerve.
In conclusion, our study [9] showed that hypertensive optic neuropathy is essentially the result of ischemia. Repeated measurements of intracranial pressure failed to show an increase in intracranial pressure in these animals with optic disc edema or optic atrophy. Thus, hypertensive optic neuropathy appears to represent one form of ischemic optic neuropathy.
Pathogenesis of Optic Disc Edema in Malignant Hypertension
There is an extensive literature on this subject; the various theories can be classified into four main categories. All these categories and their controversies are discussed at length elsewhere [8]. Briefly:
1.
Optic Disc Edema Due to Raised Intracranial Pressure: I measured the cerebrospinal fluid pressure in the monkeys with optic disc edema, and it was always normal. That rules out that theory.
2.
Optic Disc Edema Similar to Hypertensive Encephalopathy: There was no evidence of hypertensive encephalopathy in these monkeys. As discussed in detail [8], the mechanisms of hypertensive encephalopathy and hypertensive optic neuropathy are different in nature.
3.
Optic Disc Edema Part of Hypertensive Retinopathy: Since the main source of blood supply to the optic nerve head is the posterior ciliary artery circulation (see Chap. 5) and not the retinal arterial circulation, hypertensive optic neuropathy cannot be considered as a part of hypertensive retinopathy – the two are independent manifestations of malignant hypertension. Also, hypertensive retinopathy appeared significantly (p < 0.01) earlier than hypertensive optic neuropathy or choroidopathy (Fig. 23.4) [6].
4.
Optic Disc Edema Ischemic in Nature: A study of the pattern of evolution of the optic disc edema showed that in about 2–3 months, it changed to optic disc pallor; that is typically like what I have observed in patients with anterior ischemic optic neuropathy. As discussed above, light and electron microscopic studies in these animals’ eyes clearly demonstrated the ischemic nature of the hypertensive optic neuropathy [9]. Thus, the clinical and pathologic findings of the optic nerve head in this study showed that hypertensive optic neuropathy is ischemic in nature. Naturally, the question arises, what is the mechanism responsible for ischemia of the optic nerve head in these animals?
As discussed above concerning possible mechanism(s) of renovascular malignant hypertension, the available evidence indicates that the renin-angiotensin-aldosterone system plays an important role in the development and maintenance of malignant arterial hypertension. Angiotensin II (liberated by the action of renin on angiotensinogen) is a powerful vasopressor. Since choriocapillaris are very leaky, the plasma, along with angiotensin in it, leaks freely into the choroidal interstitial fluid; there it causes the vasoconstriction and/or occlusion of the choroidal vessels which was seen clearly on fluorescein fundus angiography [8] (Figs. 23.31b and 23.34) and in pathologic [9] studies in all these eyes (see “hypertensive choroidopathy” above). Moreover, ischemic choroidal lesions (see “hypertensive choroidopathy” above) were almost a universal finding in these eyes [8, 10]. The leakage of the angiotensin into the choroidal interstitial fluid and vasoconstriction and occlusion of the choroidal vascular bed would produce ischemia of the optic nerve head by two mechanisms:
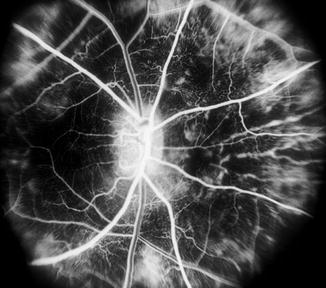
Fig. 23.34
Fluorescein angiogram during retinal arteriovenous phase in the right eye of a monkey with BP 170 mmHg shows marked choroidal filling defect (Reproduced from Hayreh et al. [8])
1.
As a part of the choroidal vascular involvement, the peripapillary choroid is also involved (Fig. 23.34). The peripapillary choroid is the main source of blood supply to the optic nerve head (Fig. 23.3) (see Chap. 5); vasoconstriction and occlusion of the peripapillary choroid would secondarily cause ischemia of the optic nerve head. There was no significant difference between the time of onset of hypertensive choroidopathy and hypertensive optic neuropathy in this study (Fig. 23.4).
2.
In addition, the angiotensin from the choroidal interstitial fluid would diffuse into the optic nerve head through the border tissue of Elschnig which is freely permeable (Fig. 23.3). The angiotensin in the tissues of the optic nerve would produce vasoconstriction and occlusion by direct action on the capillaries and other vessels in the optic nerve head; this was also suggested by the experimental studies of Sossi and Anderson [84]. Pathologic studies [9] in eyes of my study showed that the lumen of the pial vessels that supply the retrolaminar optic nerve was obliterated and that region had more marked ischemia because of this leakage of angiotensin into the retrolaminar optic nerve head as well.
Our pathologic studies in these eyes revealed that optic disc swelling was essentially due to ischemic hydropic swelling of the axons [9]. Thus, our studies strongly indicate that the optic disc edema (swelling) in malignant arterial hypertension is due to ischemia. This has been suggested by other authors [5, 63, 85] though their hypotheses about its mechanism were very different. Ischemic damage to the axons in the optic nerve head finally results in optic atrophy, which was a common finding in my animals.
In any consideration of the effect of ischemia on the neural tissue, it is essential to appreciate the fact that neural ischemia is not an “all-or-none” phenomenon. There is a whole spectrum of ischemia, varying from very mild to severe: the former producing only subtle physiological derangement of some of the neural components, while the latter results in immediate complete infarction of all neural tissues. Moreover, there is a good deal of available evidence that different components of neural tissues have a different susceptibility to ischemic damage. The subject was discussed at length elsewhere [86]. Briefly, in anterior ischemic optic neuropathy, one end of the spectrum is very mild ischemia, which in the optic nerve head produces only axoplasmic flow stasis (consequently optic disc swelling, i.e., “incipient non-arteritic anterior ischemic optic neuropathy” [87]), without interfering with the transmission of a visual impulse along the axon (hence no visual loss), while at the other extreme is the severe ischemia that causes infarction of all the neural tissues in the optic nerve head (with swollen optic disc and complete loss of vision, as in arteritic anterior ischemic optic neuropathy – see Chap. 23.19). There is a whole wide range between the two extremes. Different grades of ischemia in the optic nerve head have different effects and manifestations, and the clinical entity “anterior ischemic optic neuropathy” encompasses the entire range. In malignant hypertension, the ischemic damage to the optic nerve head may vary widely with different sequelae so that hypertensive optic neuropathy in different eyes would manifest in different ways.
Dangers of Precipitous Reduction of BP on Hypertensive Optic Neuropathy
In a discussion of hypertensive optic neuropathy, from the clinical management point of view, it is important to stress the dangers to the optic nerve head of a rapid therapeutic reduction of BP in such patients. There are number of reports [88–92] in which patients with malignant hypertension, who had their BP lowered precipitously to prevent severe neurological and cardiovascular complications, suffered immediate and permanent blindness or severe visual loss. In all these cases, the optic nerve head suffered ischemic damage and later developed marked optic atrophy. The findings were of severe bilateral anterior ischemic optic neuropathy in all cases. In addition, these patients may develop acute ischemic neurologic lesions [90–93]. It has been postulated that under such circumstances, acute ischemia of the brain, spinal cord, and/or optic nerve head is due to failure of autoregulation of blood flow in the respective organs at low BP. In the cerebral blood flow of patients with malignant hypertension of some duration, as an adaptive phenomenon, the range of the autoregulation shifts to a higher level than in normal persons (Fig. 23.1) [20, 21, 29, 94–99]. As discussed above, this adaptation makes the individual less tolerant to low BP, while at the same time it improves his tolerance of high BP. As discussed above, the optic nerve head in patients with malignant hypertension and very high BP is already ischemic, due to various microvascular changes; any sudden reduction of BP under such conditions inflicts further ischemic insult and makes the optic nerve head especially vulnerable to severe acute ischemic damage. This results in bilateral ischemia of the optic nerve head. In view of this, the BP of patients with malignant hypertension should be lowered slowly, over many hours or even days, giving time for the autoregulation of the blood flow to adapt itself to the falling BP.
Conclusion
From the evidence of my experimental study, it can be concluded that in malignant arterial hypertension, optic disc changes represent hypertensive optic neuropathy, which is a distinct entity and not simply a manifestation of either raised intracranial pressure or hypertensive encephalopathy or hypertensive retinopathy, and that hypertensive optic neuropathy is ischemic in nature and represents a form of anterior ischemic optic neuropathy. The mechanism of optic disc edema in this condition is very different from that seen with raised intracranial pressure [100]. A caution is given against a precipitous reduction of BP in patients with hypertensive optic neuropathy, because that may cause complete, permanent blindness.
Hypertensive Retinopathy
I did a comprehensive investigation of this in my experimental study with malignant arterial hypertension in rhesus monkeys. The findings of this study are discussed at length elsewhere [7, 13–17]. Following is a brief account. The retinal lesions that make up hypertensive retinopathy can be divided for descriptive purposes into the following vascular and extravascular retinal lesions, although in some of the latter, the primary factor may be retinal vascular derangement [13].
A.
Get Clinical Tree app for offline access

Retinal vascular lesions:
1.




Retinal arteriolar changes [14]
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree