Antioxidants and Age-Related Macular Degeneration: Clinical Associations and Basic Mechanisms
Yan Chen
Paul Sternberg Jr.
INTRODUCTION
Age-related macular degeneration (AMD), like many other chronic degenerative diseases, is a multifactorial disease involving environmental, genetic, and sometimes concomitant systemic disease. These risk factors impact a physiologic state of tissue and cells known as oxidative stress, which is defined as an imbalance of reactive oxygen species (ROS) and the protective antioxidant system. With aging, a decline in the retinal antioxidant defense and an increase in ROS production may shift the balance toward oxidative damage. The cellular and molecular mechanisms of retinal oxidative injury and protection, and their associations with chronic inflammation in AMD, have not yet been clearly defined. Nonetheless, there is a great body of evidence supporting the causative role of oxidative stress in the pathogenesis of AMD, and clinical studies of antioxidant supplementation have demonstrated beneficial effects in slowing the progression of AMD.
In this chapter, we review our current understanding of the role of oxidative stress in AMD based on the results of both patient-based and animal model studies. We address the experimental evidence supporting potential mechanisms of oxidative injury in the pathogenesis of AMD. We also discuss the use of antioxidants and other related novel therapeutic options in the prevention and treatment of AMD.
OXIDATIVE STRESS IN THE PATHOGENESIS OF AMD
During the past few years, tremendous progress has been made in deciphering the etiology of AMD. Major genetic variations of AMD have been mapped to genes encoding complement factor proteins and chromosome region 10q26 (1,2,3,4,5,6,7,8,9,10). Single nucleotide polymorphisms in the complement factor H (CFH) gene have been detected in a high percentage of AMD cases (1,2,3,4). However, animal models with either CFH knockout or replacement with human risk alleles have only produced minor signs of accelerated aging in the outer retina (11,12). The discrepancy between the predominant effects of genetic variations predicted by the clinical association studies and the mild phenotypic
expression in the animal models suggest that other undefined mechanisms are involved in the initiation process of macular degeneration. Considerable evidence implicates inflammatory processes as participants. Complement proteins and other proinflammatory factors may greatly amplify the initial lesions of AMD in the retinal pigment epithelium (RPE), Bruch’s membrane, and choroid. Accumulating evidence suggests that oxidative stress can be an initiating factor in the pathogenesis of AMD.
expression in the animal models suggest that other undefined mechanisms are involved in the initiation process of macular degeneration. Considerable evidence implicates inflammatory processes as participants. Complement proteins and other proinflammatory factors may greatly amplify the initial lesions of AMD in the retinal pigment epithelium (RPE), Bruch’s membrane, and choroid. Accumulating evidence suggests that oxidative stress can be an initiating factor in the pathogenesis of AMD.
The Retina Is Subjected to Oxidative Stress
As noted, oxidative stress occurs when the ROS exceeds the antioxidant capacity at the tissue and cellular level. ROS is divided into two groups based on the presence or absence of unpaired electrons: (a) Free radicals, such as superoxide anion (O2−) and hydroxyl radical (OH•), have unpaired electrons, and (b) nonradicals, such as hydrogen peroxide (H2O2), have only paired electrons. Normal physiologic processes generate ROS as by-products. For example, mitochondria produce most of the energy required by the cell via oxidative phosphorylation. During this process, around 1% to 4% of electrons leak from the electron transport chain and form O2− (13). O2− quickly converts to H2O2, which can be further transformed into highly reactive OH· in the presence of transition metals via the Fenton reaction (14,15).
Unquenched ROS readily reacts with macromolecules including proteins, lipids, and nucleic acids, which can lead to oxidative damage. To counteract the deleterious effects of ROS, cells use multilayered antioxidant systems to promptly neutralize reactive intermediates. Two types of antioxidant mechanisms serve the purpose: enzymatic detoxification systems and nonenzymatic compounds. Enzymatic antioxidants work in a coordinated manner to remove intracellular ROS. This is exemplified by the decomposition of superoxide anions (Fig. 14.1), which occurs in two sequential steps. First, superoxide dismutase (SOD) facilitates the conversion of O2− to H2O2 (16). Second, three different enzymes, catalase, glutathione peroxidase (Gpx), and peroxiredoxin (Prx), catalyze the clearance of H2O2. Mammalian catalase degrades H2O2 to water via heme groups in its active center (17). Reduction of H2O2 by Gpx occurs in the expense of reduced glutathione (GSH). In the reaction, GSH provides electrons and is oxidized to form glutathione disulfide (GSSG), and GSSG is reduced back by glutathione reductase (GR), which uses NADPH as the ultimate electron donor (18,19). The Prx system consists of three enzymes: Prx, thioredoxin (Trx), and thioredoxin reductase (TR). Prx reduces peroxides and is regenerated by Trx in conjunction with TR (20,21). Given the synchronized manner by which antioxidant enzymes work, it is easy to understand why correcting a dysfunction in a single component would often be insufficient to achieve the optimal protective effects (22,23,24).
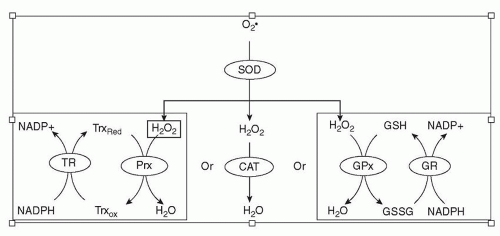 Figure 14.1 ▪ Diagrammatic representation of detoxification of ROS superoxide anion. In the presence of SOD, superoxide anion is immediately transformed into hydrogen peroxide. Three enzymes catalyze decomposition of hydrogen peroxide: catalase (CAT), Gpx, and Prx. Reduced glutathione (GSH) and Trx (TrxRed) provide electrons for Gpx and Prx, respectively. Using NADPH as the final electron donor, oxidized glutathione (GSSG) and Trx are reduced back by GR and TR. |
Nonenzymatic compounds are also key components of the antioxidant network. The tripeptide GSH is one of several highly important small compounds. It contains a free sulfhydryl (SH) group, which is highly reactive with ROS. In RPE cells, the intercellular concentration of GSH is in the millimolar range and provides a first-line buffer system against free radical attack. GSH also serves as a cofactor in the detoxification of ROS by antioxidant enzymes. Several other small molecule antioxidants, including vitamin C, vitamin E, and zinc, are also of particular importance in protecting the retina from oxidative stress. They are discussed in more detail later in the chapter.
The antioxidant systems in RPE cells are highly efficient and highly error resistant under normal physiologic conditions. However, the balance between the generation of ROS and their clearance by the antioxidant systems can be disturbed by exogenous factors, such as pollutants and tobacco use, or physiologic conditions, such as aging and concomitant systemic disease. The retina is prone
to oxidative stress for several reasons. First, the rate of oxygen consumption in the retina is greater than in any other tissue of the human body (25). This high-energy utilization is necessarily accompanied by high ROS production.
to oxidative stress for several reasons. First, the rate of oxygen consumption in the retina is greater than in any other tissue of the human body (25). This high-energy utilization is necessarily accompanied by high ROS production.
Second, the retina is continually subjected to intense focal light exposure. In the presence of photosensitizers or chromophores (visual pigments and lipofuscin), photooxidation occurs resulting in the production of ROS, which includes singlet oxygen, superoxide, hydrogen peroxide, and hydroxyl radicals (26). Third, RPE cells can function as phagocytes, phagocytizing and degrading shed photoreceptor outer segments (POS). The phagocytosis of POS stimulates respiratory burst (rapid release of ROS), which likely occurs by utilizing plasma membrane NADPH oxidase (27,28,29). Moreover, the outer segments are extremely abundant in polyunsaturated fatty acids (PUFAs), which readily undergo lipid peroxidation. The resultant oxidized intermediates are long-lived and covalently modify various kinds of molecules including proteins, thereby impairing the stability and functions of their targets. Thus, lipid peroxidation initiates a chain reaction, which amplifies the initial number of free radicals. Two major products of lipid peroxidation, carboxyethylpyrrole (CEP) and malondialdehyde (MDA), have been recently linked to AMD and are discussed in more detail later in the chapter.
As the retina ages, it becomes more susceptible to oxidative injury due partly to an age-related decline in the antioxidant capacity. For example, a reduction in catalase activity in aging RPE was demonstrated by both biochemical and immunohistochemical analyses of human donor eyes (30,31). Systemically, the plasma GSH pool has an age-dependent decrease in total level as well as an oxidation in the redox status (32,33). Meanwhile, age-related increases in chromophores like lipofuscin cause increases in pro-oxidants that further stress and sometimes surpass the antioxidant system capacity.
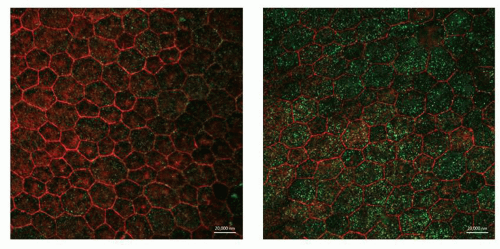 Figure 14.2 ▪ Increased lipofuscin fluorescence in aging RPE from Nrf2-deficient mice. RPE/choroid flat mount was prepared from age-matched (14 M) wild-type (WT) and Nrf2 knockout (KO) mice. Autofluorescence (green) was monitored over a broad spectrum of excitation wavelengths (e.g., 488 and 555 nm). F-actin was stained with phalloidin (red) to delineate the boundaries of the RPE. Increased autofluorescence in Nrf2-deficient RPE is an indicator of lipofuscin accumulation. Scale bar: 20 µm. |
Lipofuscin
Lipofuscin, also known as aging pigment, is an autofluorescent and electron-dense material that accumulates with age in postmitotic cells, including the RPE (34). It has a brownyellow color and is enzymatically nondegradable. The rate of lipofuscin accumulation shows a negative correlation with longevity in animals. Thus, the accumulation of lipofuscin is considered a hallmark of aging (35,36). An age-dependent buildup of lipofuscin occurs in human RPE cells; at the age of 90, lipofuscin occupies about 19% of the RPE cell volume, whereas in the first 10 years of life, it occupies only 1% (37). The increased lipofuscin in the RPE causes an increase in autofluorescence of the fundus and precedes the development of geographic atrophy. Thus, high autofluorescence of the fundus can be a useful clinical biomarker that can assist in monitoring the progression of atrophic AMD (37,38,39) (Fig. 14.2).
The composition of lipofuscin is highly heterogeneous and includes oxidized proteins and lipids. Two degradation pathways, autophagy and phagocytosis, contribute to lipofuscin accumulation in the lysosomes. In the RPE, the role of phagocytosis has been well established by multilayered evidence. On the molecular level, the best-characterized fluorophore of the RPE lipofuscin, N-retinylidene-N-retinylethanolamine (A2E), derives from unique components of phagocytized POS (40,41). On the cellular level, long-term exposure of POS leads to formation of lipofuscin-like autofluorescent granules in cultured RPE cells (42). On the whole animal level, RCS
rats (which have early photoreceptor degeneration) show less lipofuscin accumulation with age in the RPE (43). Compared to phagocytosis, the role of autophagy in RPE lipofuscin formation is less defined. However, results from both in vivo and in vitro studies indicate that RPE lipofuscin is not produced solely from phagocytosis. Studies with long-term cultured RPE cells show that autofluorescent inclusions resembling lipofuscin granules accumulate in the absence of outer segments or the A2E (44). Mice lacking alphaVbeta5 integrin are defective in phagocytosis of POS; nonetheless, lipofuscin accumulation with age is still apparent in the RPE of these mice (45). We recently developed a murine model of AMD using nuclear factor erythroid 2-related factor (Nrf2)-deficient mice. In this model, increased presence of autophagy intermediate structures was detected in the aging RPE, indicating dys-regulated autophagy (46). Lipofuscin-associated fluorescence was more pronounced in aging RPE from knockout mice than in an age-matched wild-type control. More conclusively, components originating from autophagy have been identified in human RPE lipofuscin using proteomic approaches (47,48). Thus, like in other postmitotic tissues, autophagy appears to be involved in lipofuscinogenesis in the RPE (49).
rats (which have early photoreceptor degeneration) show less lipofuscin accumulation with age in the RPE (43). Compared to phagocytosis, the role of autophagy in RPE lipofuscin formation is less defined. However, results from both in vivo and in vitro studies indicate that RPE lipofuscin is not produced solely from phagocytosis. Studies with long-term cultured RPE cells show that autofluorescent inclusions resembling lipofuscin granules accumulate in the absence of outer segments or the A2E (44). Mice lacking alphaVbeta5 integrin are defective in phagocytosis of POS; nonetheless, lipofuscin accumulation with age is still apparent in the RPE of these mice (45). We recently developed a murine model of AMD using nuclear factor erythroid 2-related factor (Nrf2)-deficient mice. In this model, increased presence of autophagy intermediate structures was detected in the aging RPE, indicating dys-regulated autophagy (46). Lipofuscin-associated fluorescence was more pronounced in aging RPE from knockout mice than in an age-matched wild-type control. More conclusively, components originating from autophagy have been identified in human RPE lipofuscin using proteomic approaches (47,48). Thus, like in other postmitotic tissues, autophagy appears to be involved in lipofuscinogenesis in the RPE (49).
Lipofuscin augments oxidative stress by increasing ROS generation and weakening antioxidant defense.
Lipofuscin granules are photosensitizers that produce ROS in response to light exposure (50,51,52,53). It has recently been reported that photo-oxidation products of the major lipofuscin fluorophore A2E may contribute to drusen formation in human AMD (54). They may also directly damage the intracellular antioxidant defense. In fact, RPE cells fed with the aging pigment have decreased activity of SOD and catalase (55). Accumulated lipofuscin compromises cellular functions including lysosome-mediated degradative pathways, which are responsible for removal of damaged proteins and organelles (55,56,57,58). Altogether, lipofuscin causes considerable oxidative damage in aging RPE by increasing ROS production and impairing removal of ROS.
ASSOCIATION BETWEEN AMD AND OXIDATIVE STRESS: EVIDENCE BASED ON EPIDEMIOLOGIC AND CLINICAL ASSOCIATION STUDIES
Population-based studies have established a strong correlation between oxidative stress and AMD. Risk factors of AMD, such as aging, smoking, and sunlight exposure, are closely linked to oxidative stress (59,60,61,62,63,64,65,66). Markers of oxidative damage are detected both locally and systemically in patients with AMD. Moreover, antioxidant supplementation is an effective intervention in delaying the progression of AMD (67). Together, epidemiologic and clinical studies clearly demonstrate an association between oxidative stress and AMD.
Oxidation Markers in the AMD
Oxidative stress can be evaluated by various markers. In the past, we used plasma thiol/disulfide redox status and/or the concentration of thiol metabolites as a systemic indicator of oxidative stress. We showed that oxidation of plasma GSH and cysteine pools is associated with AMD risk factors including aging, smoking, and CFH polymorphism (32,62,68,69). Our recent studies further showed that plasma level of oxidized cysteine is sensitive to the supplements recommended by the Age-Related Eye Disease Study (AREDS). Dietary antioxidants and zinc indeed reverse the tendency of oxidation of the plasma cysteine in patients with AMD (70,71). Therefore, monitoring the redox status of plasma cysteine may provide a tool for predicting the effectiveness of antioxidant intervention in AMD patients.
The retina is rich in PUFA and susceptible to lipid peroxidation, and therefore, lipid peroxidation has been widely studied in the context of AMD. A relationship between dietary intake of PUFA and AMD is proposed by several published studies. In human eyes with AMD, an increased staining of oxidized phospholipids is prominent in the outer segments and the RPE (72). Presence of lipid modification in lipofuscin from human RPE is confirmed by proteomic analysis (48). Drusen, the earliest clinical manifestation of AMD, contains adducts of CEP and MDA, both of which are products of lipid peroxidation (73,74). The increased level of those two adducts is observed not only in the RPE/Bruch’s membrane/choroid complex but also in the plasma of patients with AMD (74,75,76). Recent studies on those two lipid peroxidation markers have provided critical information in elucidating functional roles for oxidative stress in AMD.
CEP adducts are derived from oxidation of docosahexaenoic acid (DHA), a fatty acid component of the membrane. DHA is an essential PUFA with highest abundance in the retina and accounts for approximately 50% of the fatty acids in POS membrane (77). Oxidative cleavage of DHA gives rise to 4-hydroxy-7-oxohept-5-enoic acid, and the latter covalently modifies proteins to form CEP adducts (78). Immunocytochemical studies showed that in the eye, CEP adducts mainly locate in the POS and RPE (76). CEP modifications have been identified in drusen of human AMD eyes. In addition, human AMD eyes have more CEP adducts in RPE/Bruch’s membrane/choroid complex than do age-matched controls (73). Systemically, AMD human plasma has increased levels of CEP adducts and anti-CEP autoantibodies compared with age-matched non-AMD control human plasma (76,79). CEP adducts are not only an oxidative damage marker associated with AMD. A recent study by Hollyfield et al. (80) showed that systemic administration of CEP adducts can initiate dry AMD-like phenotype in animal models. Thus, these studies convincingly suggest that the CEP adducts contribute to the AMD pathology of the outer retina.
MDA is a commonly used lipid peroxidation marker. In contrast to CEP (which is uniquely generated from DHA), MDA is the decomposition product from oxidation of PUFAs
that contain more than two methylene-interrupted double bonds (81). Plasma samples from patients with AMD have higher levels of MDA than that of a control cohort (82,83). The negative regulator of the complement system, CFH, was recently found to specifically bind to the plasma MDA adducts, but not to the other oxidative modifications tested, such as CEP and 4-hydroxynonenal (74). CFH binds to MDA adducts via its two short consensus repeats (SCRs), SCR7 or SCR20, and neutralizes proinflammatory effects of MDA adducts. More strikingly, the AMD risk allele of CFH Y402H, which has an amino acid substitution in the region of SCR7, has impaired binding ability to MDA modifications and decreased protection. Together, using both clinical and experimental approaches, this elaborate study provides the first clue to solve the mystery on functional roles of the genetic risk factor CFH in AMD.
that contain more than two methylene-interrupted double bonds (81). Plasma samples from patients with AMD have higher levels of MDA than that of a control cohort (82,83). The negative regulator of the complement system, CFH, was recently found to specifically bind to the plasma MDA adducts, but not to the other oxidative modifications tested, such as CEP and 4-hydroxynonenal (74). CFH binds to MDA adducts via its two short consensus repeats (SCRs), SCR7 or SCR20, and neutralizes proinflammatory effects of MDA adducts. More strikingly, the AMD risk allele of CFH Y402H, which has an amino acid substitution in the region of SCR7, has impaired binding ability to MDA modifications and decreased protection. Together, using both clinical and experimental approaches, this elaborate study provides the first clue to solve the mystery on functional roles of the genetic risk factor CFH in AMD.
CAUSAL ROLES OF OXIDATIVE STRESS IN AMD: EVIDENCE FROM ANIMAL MODELS
Despite decades of efforts and the strong clinical association between oxidative stress and AMD, mechanisms of how oxidative injury leads to RPE degeneration and the link with chronic inflammation remain elusive. Severe oxidative stress induces apoptosis, but loss of RPE cells occurs mainly in the late stages of AMD. Cellular and molecular mechanisms of the early RPE degeneration remain largely unknown. In recent years, compelling evidence has been obtained from animal models, in which oxidative stress is introduced either by increasing exogenous pro-oxidant level, such as injection of CEP, or by compromising endogenous antioxidant systems such as deletion of the stress response gene Nrf2.
Superoxide Dismutase Knockout Mice
Mammals have three isoforms of SOD: the cytoplasmic Cu/Zn-SOD (SOD1), the mitochondrial MnSOD (SOD2), and the extracellular Cu/Zn-SOD (SOD3). All three require catalytic metals (Cu or Mn) for their activities (16). SODs play significant roles in oxidative defense, which is evident by increased oxidative damage in mice deficient of the cytoplasmic SOD1 or the mitochondrial SOD2. In fact, mice that completely lack the SOD1 have accelerated aging and shortened lifespan, and SOD2 knockout mice are lethal (84,85).
SODs are highly expressed in the retina (86), and manipulation of SODs in mice generates phenotypes related to human AMD. Mice deficient of the cytoplasmic SOD1 are the first animal model that demonstrated the causative roles of oxidative stress in pathogenesis of AMD (87). The SOD1 knockout mice showed AMD-like pathology in the outer retina (deposition of inflammatory proteins in the RPE/choroid complex, thickened Bruch’s membrane, and sub-RPE deposition) and developed drusen after 9 months. In young knockouts, constant light exposure at an intensity similar to outdoor sunlight caused drusen formation within a few weeks. After 10 months, about 10% of the mice developed choroidal neovascularization (CNV) and subretinal fluid leakage during fluorescein angiography. SOD2 is critical in defense against mitochondrial oxidative stress. Sod2−/− mice have dilated cardiomyopathy and die within weeks after birth (85). Retinal toxicity is evident even in these short-lived mutant mice, which show a progressive thinning of retina mainly involving the inner retinal and photoreceptor layers (88). RPE-specific knockdown of SOD2 leads to phenotypic changes related to AMD, but mainly in the RPE/Bruch’s membrane. Those changes include accumulation of oxidized proteins in the RPE/choroid complex, vacuolization and degeneration of the RPE, thickening of Bruch’s membrane, and increased levels of autofluorescence and A2E (89).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


