Abnormalities of the Posterior Segment
Pamela Hooker
Ocular pathology in the posterior segment is relatively uncommon in children. When it does occur, however, it can be serious, with vision- and even life-threatening consequences. Clinicians must perform careful examinations of the posterior segment of all of their pediatric patients, including regular dilated fundus examinations. The most frequently encountered conditions will be discussed, along with current treatments and referral options.
Ciliary Body
Posterior Uveitis
Posterior uveitis in the pediatric population has many causes. Among these are toxoplasmosis, toxocariasis, sarcoidosis, bacterial infections (e.g., tuberculosis and syphilis), viral infections (e.g., cytomegalovirus [CMV], rubella, measles, and herpes viruses), and fungal infections (1). Symptoms can include blurred vision, redness, and floaters, but usually no pain. Treatment varies according to the causative agent, but often includes systemic steroids (2).
In older children, toxoplasmosis is the most common cause of posterior uveitis (Fig. 13.1). It is caused by Toxoplasma gondii and is carried by cats. It can be congenital or acquired, although chorioretinitis is usually associated with the former. Lesions appear fluffy and yellow-white in active cases and are usually located adjacent to an area of chorioretinal atrophy or pigmented scar. Often, there is an overlying vitritis. Macular involvement is usually seen, with significant visual acuity impairment (3), but toxoplasmosis cases are often self-limiting, resolving within weeks to months. In the case of an immunocompromised patient, however, more severe ocular disease can occur (4). When treatment is indicated, as in the case of lesions close to the optic nerve or macula, or in immunocompromised patients, it is usually in the form of pyrimethamine or clindamycin and steroids.
Toxocariasis is caused by infection with the intestinal parasite Toxocara canis, common in dogs. It can present as endophthalmitis, retinal granulomas (especially in the macula), or more widespread inflammation with the possibility of vitreoretinal abscess (1,5). The condition is generally unilateral. As with toxoplasmosis, systemic steroids are indicated to reduce inflammation. The use of antihelmintic drugs may be counterproductive because the death of the parasite can cause additional inflammation.
Posterior uveitis caused by systemic sarcoid is more common in patients between the ages of 20 and 50 years, but is occasionally seen in younger patients. Typical findings include bilateral pain and photophobia, as well as blurred vision. Severe cases may show mutton-fat keratic precipitates and iris nodules. Systemic steroids are indicated because topical steroids will be insufficient.
Bacterial infections (tuberculosis, syphilis) and viral infections (CMV, rubella, measles,
herpes zoster, herpes simplex) can also cause pediatric posterior uveitis. Approximately 6% of children with the acquired immunodeficiency syndrome (AIDS) will have CMV retinitis (1), which appears as a whitish necrotic area in the retina with associated retinal hemorrhages (5). Intravenous ganciclovir should be administered. Congenital syphilis presents as a salt-and-pepper fundus and can mimic any number of other conditions. In any case of suspected viral or bacterial infection, workup should include appropriate testing to rule out probable organisms so that the proper antibiotic or other treatment can be administered.
herpes zoster, herpes simplex) can also cause pediatric posterior uveitis. Approximately 6% of children with the acquired immunodeficiency syndrome (AIDS) will have CMV retinitis (1), which appears as a whitish necrotic area in the retina with associated retinal hemorrhages (5). Intravenous ganciclovir should be administered. Congenital syphilis presents as a salt-and-pepper fundus and can mimic any number of other conditions. In any case of suspected viral or bacterial infection, workup should include appropriate testing to rule out probable organisms so that the proper antibiotic or other treatment can be administered.
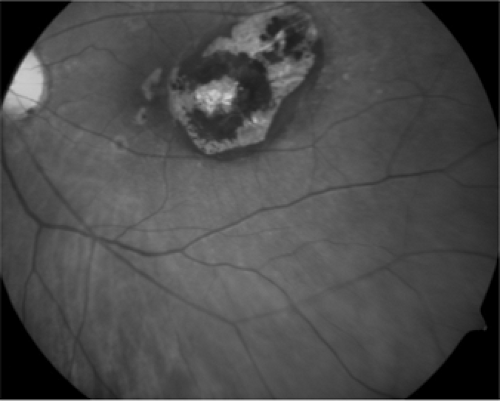 Figure 13.1. Toxoplasmosis (photo courtesy of Dr. Jeffrey Roth). (See color well.) |
Panuveitis
Widespread uveitis can occur secondary to many of the above conditions if not treated promptly, as well from such entities as Behçet disease. The latter is diagnosed based on a combination of oral ulcers plus two of a number of systemic manifestations, including bilateral retinal vasculitis (6,7,8). Although the disease is more common in patients from 20 to 50 years of age, when a pediatric onset occurs, uveitis will more likely be one of the presenting symptoms (9). As with other cases of posterior uveitis, symptoms include blurry vision, photophobia, pain, and floaters. Complications are frequent and include hypopyon, cataract, macular edema, retinal vascular changes including branch retinal vein occlusion (BRVO) and neovascularization, and retinal detachment (1,5). These children will need systemic steroids and referral to a specialist for possible immunosuppressive therapy.
Vitreous
Persistent Hyperplastic Primary Vitreous
A significant developmental disorder of the vitreous is persistent hyperplastic primary vitreous (PHPV). Occasionally, the hyaloid vascular system will not be completely regressed at the time of an infant’s birth. On examination it may appear as a projection of tissue from the optic disk or as a small opacity on the posterior aspect of the crystalline lens known as a Mittendorf dot (Fig. 13.2). More significant is the failure of proper development of the primary vitreous. When PHPV is present, it is usually associated with microphthalmia, shallow anterior chamber, lens opacities, and retrolenticular membrane, and it can present with leukocoria of the affected eye or retinal detachment (10,11,12). The condition may progress and cause angle-closure glaucoma. Treatment varies with the age of onset, but can include lensectomy or vitrectomy, with accompanying occlusion therapy. Contact lens fitting is indicated to manage the aphakic refractive error.
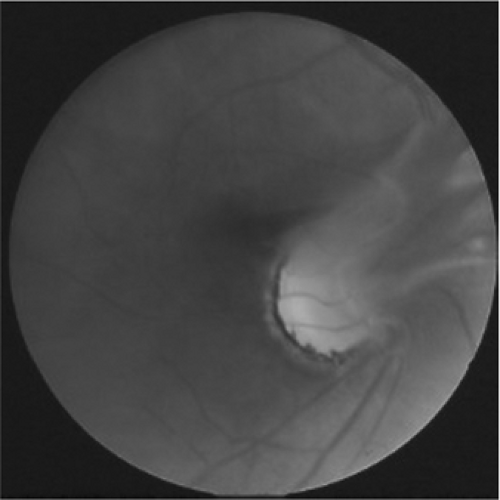 Figure 13.2. Persistent hyperplastic primary vitreous (PHPV) (photo courtesy of Dr. Scott Richter). (See color well.) |
Retina
Developmental Anomalies
Coloboma
Colobomas can occur in many ocular structures, including the iris, optic nerve, and retina (Fig. 13.3). Coloboma, a developmental anomaly that represents incomplete closure of the fetal fissure during gestational development, can be unilateral or bilateral. Usually, no family history of coloboma is found, but an autosomal dominant inheritance pattern has been shown in some cases.
Retinal or choroidal colobomas typically appear as a pale yellow-white area inferior to the optic disk, and may present as leukocoria if sufficiently large. They vary in size and can be associated with optic nerve coloboma. If this is the case, retinal detachment is also a concern. Visual prognosis is closely associated with the amount of macular and optic nerve involvement. An associated nonprogressive visual field defect may also be present. Associated systemic abnormalities include CHARGE association, basal encephalocele, and various chromosomal disorders (13,14).
Optic pits and morning glory syndrome have been described as mild and severe presentations of optic nerve coloboma, respectively (15). The latter involves a posteriorly displaced optic nerve head within a posterior staphyloma, with several layers of glial tissue covering it. The retinal vessels radiate outward like spokes on a wheel, and vision is generally poor from birth.
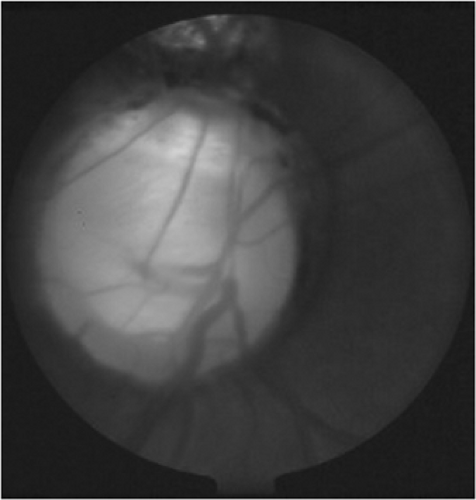 Figure 13.3. Optic nerve head coloboma (photo courtesy of Dr. Scott Richter). (See color well.) |
Vascular Anomalies
Capillary Hemangioma
Capillary hemangiomas in the retina typically occur as part of Von Hippel-Lindau disease (16), although they can sometimes be seen in isolation (17). They appear as elevated, red vascular lesions that vary in size and have feeder and drainage vessels. They can occur either in the retina or on the optic disk, the latter of which are more difficult to treat. By themselves, capillary hemangiomas are benign tumors, but they can cause blurred vision or vision loss from retinal exudation, epiretinal membrane formation, or vitreous hemorrhage. Treatment usually consists of either cryotherapy or argon laser, with generally good visual outcomes (18,19).
Cavernous Hemangioma
Cavernous hemangiomas typically appear in females in the second or third decade of life. They have a grape cluster appearance, with small aneurisms surrounded by gray-white fibrous tissue, and are usually unilateral and isolated. Most cavernous hemangiomas can be observed unless leakage is evident (14,20).
Arteriovenous (AV) Malformation
Although most vascular abnormalities in children are benign when they occur in isolation, clinicians should be aware that some can be associated with systemic disease. Diabetes can cause venous beading, and leukemia can cause beading or tortuosity of retinal vessels, for example. Any vascular anomaly noted in a pediatric patient should be followed up with appropriate systemic testing.
Retinopathy of Prematurity
Retinopathy of prematurity (ROP), a vasoproliferative disorder caused by incomplete retinal vascularization, is exacerbated by oxygen
exposure. Because of the increasing survival rate of premature infants, ROP is becoming more of a concern for pediatric optometrists and ophthalmologists. Peripheral vascularization reaches the nasal ora serrata at approximately 36 weeks’ gestational age, but does not reach the temporal ora until 40 weeks (21). Premature infants, therefore, do not have complete retinal vascularization at the time of birth. Infants at high risk for ROP include those who are born at less than 36 weeks’ gestation or weigh less than 2000 g, particularly if the child with highest risk associated with extremely low birth-weight (less than 1000 g), received supplemental oxygen after birth (21). The administration of oxygen causes normal vascularization to stop, and abnormal neovascularization can occur.
exposure. Because of the increasing survival rate of premature infants, ROP is becoming more of a concern for pediatric optometrists and ophthalmologists. Peripheral vascularization reaches the nasal ora serrata at approximately 36 weeks’ gestational age, but does not reach the temporal ora until 40 weeks (21). Premature infants, therefore, do not have complete retinal vascularization at the time of birth. Infants at high risk for ROP include those who are born at less than 36 weeks’ gestation or weigh less than 2000 g, particularly if the child with highest risk associated with extremely low birth-weight (less than 1000 g), received supplemental oxygen after birth (21). The administration of oxygen causes normal vascularization to stop, and abnormal neovascularization can occur.
According to the International Classification of Retinopathy of Prematurity (22), the disease is categorized into five stages in three retinal zones. Zone I is defined as a circle with the optic disk at the center and a diameter of twice the distance from the optic nerve to the fovea. Zone II is a circle concentric to Zone I that extends outward to the nasal ora serrata. Zone III is the remaining retina, a crescent including the superior, inferior, and temporal periphery.
Stage I disease consists of a demarcation line between vascular and avascular retina in the temporal periphery. In Stage II, this line is elevated into a ridge. Stage III disease includes neovascular vessels extending into the vitreous from this area. If retinal detachment exists, the ROP is at least Stage IV, with Stage IV-a having the macula attached and Stage IV-b having the macula detached. Total retinal detachment is seen in Stage V.
Three further classifications of ROP are plus disease, rush disease, and threshold disease. In plus disease, dilated and tortuous retinal vessels are seen in combination with dilated iris vessels (Fig. 13.4). Rush disease describes the involvement of Zone I with extensive disease. Threshold disease is reached when the child has Stage III ROP, in Zones I or II, with disease that includes either five contiguous or eight total clock hours of neovascularization.
Progression of ROP to disease is the indication for treatment, which usually consists of either cryotherapy to the avascular peripheral retina or, more recently, laser photocoagulation. Cryotherapy has been shown to reduce the incidence of unfavorable outcomes in ROP by 50% (21,23). Additional surgery may be indicated for retinal detachment.
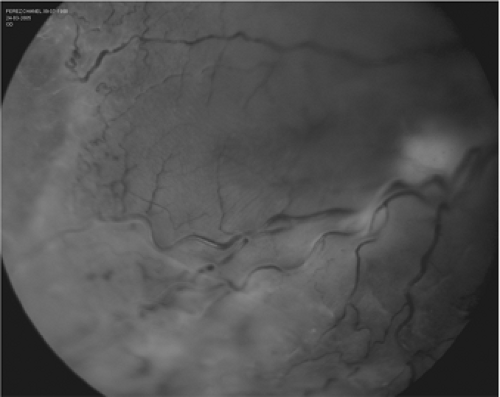 Figure 13.4. Retinopathy of prematurity (ROP) with retinal detachment (photo courtesy of Dr. Jeffrey Roth). (See color well.) |
Genetic Disorders
Congenital Stationary Night Blindness
Congenital stationary night blindness (CSNB) is a genetic condition that can be inherited in an autosomal dominant, autosomal recessive, or X-linked pattern. Most patients with CSNB show either the X-linked or autosomal recessive inheritance pattern, and have nystagmus in infancy, myopia with reduction in visual acuity, and mild color vision deficiency. The autosomal dominant form does not show nystagmus and has normal visual acuity. All forms show poor night vision and reduced scotopic response on electroretinogram (ERG) (24).
Achromatopsia
Two distinct forms of achromatopsia exist: an autosomal recessive form, which is more common and also known as rod monochromatism; and an X-linked recessive form, also known as blue cone monochromatism. The autosomal recessive form, in which there is a complete lack of functional cone photoreceptors in the retina, shows absent cone responses on ERG (14), reduced vision (often with high hyperopia), rapid, fine nystagmus, and severe photophobia. In the X-linked form, presentation is milder, with
typically myopic refractive error, better visual acuity, and some blue cone function (24).
typically myopic refractive error, better visual acuity, and some blue cone function (24).
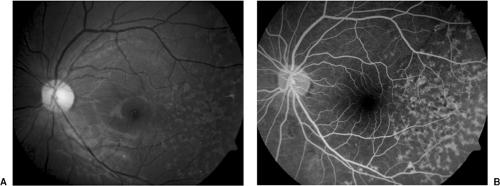 Figure 13.5. Stargardt’s disease. A: Color fundus photo (see color well). B: Fluorescein angiography (photos courtesy of Dr. Jeffrey Roth). |
Leber’s Congenital Amaurosis
Also known as infantile rod-cone dystrophy, Leber’s congenital amaurosis is an autosomal recessive inherited disorder. Children with Leber’s amaurosis generally have a normal-appearing fundus, but extremely poor vision with roving eye movements, nystagmus, and high hyperopia. Pupil responses are also abnormal and ERG response is severely decreased or extinguished. The condition is typically detected within the first few months of life, and patients suspected of having Leber’s amaurosis should be referred for systemic testing to rule out rare syndromes such as Joubert syndrome (24,25).
Stargardt Disease
Stargardt disease is an autosomal recessive disorder, which usually presents as a loss of central vision in school-aged children (26). In its early stages, the fundus may appear normal, although the typical presentation is a bull’s-eye maculopathy that is often accompanied by flecklike yellow deposits at the level of the retinal pigment epithelium (RPE) (Fig. 13.5a). (A variant of Stargardt, fundus flavimaculatus, shows these flecks without maculopathy, and has a better visual prognosis.) Fluorescein angiography shows a characteristic dark choroid and central atrophy (Fig. 13.5b). Indocyanine green angiography shows large choroidal vessels in the “dark” area and hypofluorescence of flecks (27) in contrast to the hyperfluorescence seen in fluorescein angiography. Pattern ERG is frequently reduced (28




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
