EYELID DISORDERS
Epicanthus
Summary
Epicanthus is a skin fold that extends from upper to lower eyelid, overlying the medial canthus.
Demographics
 Typical in infants of Asian ethnicity.
Typical in infants of Asian ethnicity.
 Common in others; diminishes over time with maturation of the face.
Common in others; diminishes over time with maturation of the face.
Symptoms and Signs
 Pseudoesotropia results from partial concealment of nasal sclera.
Pseudoesotropia results from partial concealment of nasal sclera.
 Most evident in partial lateral gaze, which makes abducting eye appear centered in the palpebral fissure while adducting eye appears to be “in the corner” (Figure 8.1).
Most evident in partial lateral gaze, which makes abducting eye appear centered in the palpebral fissure while adducting eye appears to be “in the corner” (Figure 8.1).

FIGURE 8.1. Pseudoesotropia caused by epicanthal folds. Slight right face turn exaggerates impression that right eye is turned inward, while left eye appears centered in the palpebral fissure.
Systemic Findings
 Many conditions involving craniofacial dysmorphism have epicanthus as a typical feature, frequently associated with additional eyelid anomaly (such as abnormal palpebral fissure slant). Foremost among these is Down syndrome (trisomy 21, the most common chromosomal disorder), in which epicanthus and “mongoloid” (nasal lower than temporal) fissure slant are consistently seen, and which can affect the eyes in many other ways.
Many conditions involving craniofacial dysmorphism have epicanthus as a typical feature, frequently associated with additional eyelid anomaly (such as abnormal palpebral fissure slant). Foremost among these is Down syndrome (trisomy 21, the most common chromosomal disorder), in which epicanthus and “mongoloid” (nasal lower than temporal) fissure slant are consistently seen, and which can affect the eyes in many other ways.
Common ocular manifestations include blepharitis, congenital nasolacrimal duct obstruction, ectropion, Brushfield spots of the iris (multiple dotlike whitish condensations of iris stroma near the pupil margin), optic disc anomalies (most commonly increased number of blood vessels), myopia, astigmatism, strabismus, and congenital nystagmus.
There is increased incidence of cataract, glaucoma, and keratoconus in childhood. Mental impairment ranges from mild to profound. Important systemic findings include cardiac malformations and upper cervical spine instability.
Although it occurs with much increased frequency in children born to older women, overall a majority of children with Down syndrome have younger mothers.
 Fetal alcohol syndrome, caused by excessive maternal alcohol consumption during pregnancy, is characterized by epicanthus, telecanthus (see below), and often ptosis, typically associated with a thin vermilion border of the upper lip and flattened philtrum. Common fundus abnormalities include mild to moderate hypoplasia or dysplasia of the optic discs, and tortuous retinal vessels. Strabismus, nystagmus, and both high hyperopia and high myopia occur with increased frequency, as do various forms of anterior segment dysgenesis (p. 440). Retardation of both physical growth and mental development to some degree is typical, and there is increased incidence of cardiac, skeletal, and other organ abnormalities.
Fetal alcohol syndrome, caused by excessive maternal alcohol consumption during pregnancy, is characterized by epicanthus, telecanthus (see below), and often ptosis, typically associated with a thin vermilion border of the upper lip and flattened philtrum. Common fundus abnormalities include mild to moderate hypoplasia or dysplasia of the optic discs, and tortuous retinal vessels. Strabismus, nystagmus, and both high hyperopia and high myopia occur with increased frequency, as do various forms of anterior segment dysgenesis (p. 440). Retardation of both physical growth and mental development to some degree is typical, and there is increased incidence of cardiac, skeletal, and other organ abnormalities.
Differential Diagnosis
 In infancy, intermittent esotropia may be infrequently manifest and not detectable on examination, leading to an erroneous diagnosis of pseudoesotropia. Care must be taken to differentiate these conditions and to avoid inappropriately dismissing parental concerns about strabismus merely because pseudoesotropia is present.
In infancy, intermittent esotropia may be infrequently manifest and not detectable on examination, leading to an erroneous diagnosis of pseudoesotropia. Care must be taken to differentiate these conditions and to avoid inappropriately dismissing parental concerns about strabismus merely because pseudoesotropia is present.
Family photos believed to show esodeviation, especially when eyes are captured in partial lateral gaze (often evident from head posture that partially conceals one ear), may be used to confirm suspicion of pseudoesotropia and clarify the distinction for parents.
Parental description of unilateral convergence movement (one eye “pulls in” momentarily at times) is strongly suggestive of true intermittent esotropia, especially when seen mainly in association with fatigue or sleepiness.
Pseudoesotropia never gets worse. Family should be strongly cautioned to bring the child back for reevaluation if there is increasing perception of strabismus.
 Telecanthus is increased distance between the medial canthi, with normal interpupillary distance. (Much less common is true hypertelorism, or increased interpupillary distance.) Epicanthal folds, often associated, must be retracted by the examiner to make this determination. Normal intermedial canthal distance is approximately equal to the horizontal width of the palpebral fissure.
Telecanthus is increased distance between the medial canthi, with normal interpupillary distance. (Much less common is true hypertelorism, or increased interpupillary distance.) Epicanthal folds, often associated, must be retracted by the examiner to make this determination. Normal intermedial canthal distance is approximately equal to the horizontal width of the palpebral fissure.
Telecanthus is a typical finding in Waardenburg syndrome, also characterized by laterally displaced lacrimal puncta, white forelock, decreased iris stromal pigmentation (often unilateral, causing heterochromia), and congenital sensorineural deafness (in about 20% of cases).
Cornelia de Lange syndrome also usually includes telecanthus, though synophrys (“unibrow”) is the most typical facial feature (seen as well in some cases of Waardenburg). Other ocular findings include early-onset myopia, lacrimal drainage obstruction, blepharitis, ptosis, strabismus, and nystagmus. Gastroesophageal reflux and small or absent digits are characteristic, as is the presence of moderate-severe cognitive and behavioral problems and variable hearing impairment.
Abnormal Lid Apposition
Table 8.1 summarizes a number of conditions in which there is congenital or acquired disturbance of the relationship between the eyelids and/or lashes and the ocular surface. Young corneas usually are quite tolerant to moderate degrees of exposure (Figure 8.2) and contact with (relatively soft) lashes; these disorders often cause no symptoms or only mild ones, and seldom lead to scarring unless there is significant comorbidity, such as tear insufficiency.
Table 8.1 Causes of faulty eyelid apposition in childhood
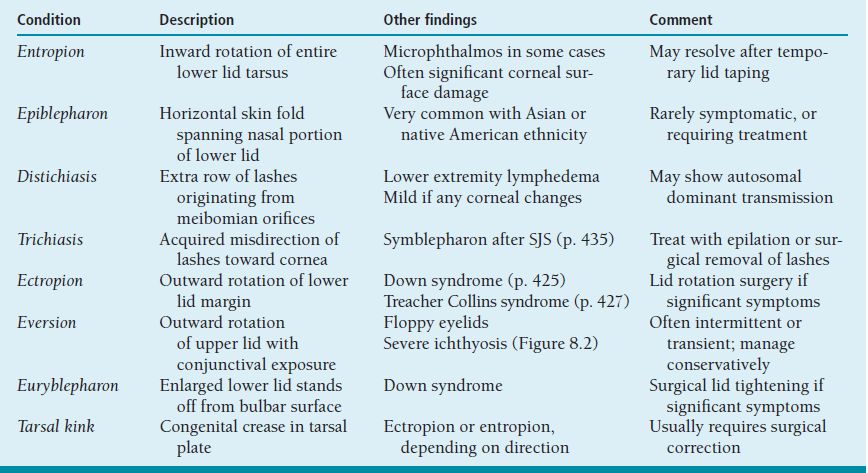
FIGURE 8.2 Eversion of upper eyelids in a newborn with lamellar ichthyosis (“collodion baby”). Skin condition improved and lids returned to normal position over several months.
Eyelid Coloboma
Summary
Eyelid coloboma is congenital absence of a portion of the lid, including margin.
Ophthalmic Findings
 Varies from small notch to absence of entire lid, usually upper.
Varies from small notch to absence of entire lid, usually upper.
 Absent tarsus, lashes, and meibomian glands.
Absent tarsus, lashes, and meibomian glands.
 If the defect is large, exposure keratitis may quickly become a serious problem.
If the defect is large, exposure keratitis may quickly become a serious problem.
Systemic Findings
 Goldenhar syndrome (hemifacial microsomia; oculoauriculovertebral dysplasia) includes upper lid coloboma in 20% of cases. Epibulbar dermoid involving limbus or conjunctiva is the most common ocular abnormality (p. 474). Duane syndrome is also a frequent eye finding (p. 508). Other craniofacial anomalies include external (tags, microtia) and internal ear malformation (often with hearing loss), maxillary and mandibular hypoplasia, and macrostomia. Vertebral and cardiac abnormalities may be present. Findings are usually unilateral; sporadic (nonfamilial) occurrence.
Goldenhar syndrome (hemifacial microsomia; oculoauriculovertebral dysplasia) includes upper lid coloboma in 20% of cases. Epibulbar dermoid involving limbus or conjunctiva is the most common ocular abnormality (p. 474). Duane syndrome is also a frequent eye finding (p. 508). Other craniofacial anomalies include external (tags, microtia) and internal ear malformation (often with hearing loss), maxillary and mandibular hypoplasia, and macrostomia. Vertebral and cardiac abnormalities may be present. Findings are usually unilateral; sporadic (nonfamilial) occurrence.
 Treacher Collins syndrome (mandibulofacial dysostosis) typically includes temporal lower lid coloboma, with “antimongoloid” (nasal higher than temporal) slant of palpebral fissures. Malar and mandibular hypoplasia, ear malformations (often with deafness), and macrostomia are also characteristic findings. Usually bilateral; autosomal dominant inheritance, but most cases represent new mutations.
Treacher Collins syndrome (mandibulofacial dysostosis) typically includes temporal lower lid coloboma, with “antimongoloid” (nasal higher than temporal) slant of palpebral fissures. Malar and mandibular hypoplasia, ear malformations (often with deafness), and macrostomia are also characteristic findings. Usually bilateral; autosomal dominant inheritance, but most cases represent new mutations.
 Amniotic band syndrome may cause eyelid coloboma. Other findings include facial clefts, limb malformation or amputation; patterns variable and not consistent with interruption of normal developmental sequence. Typically sporadic.
Amniotic band syndrome may cause eyelid coloboma. Other findings include facial clefts, limb malformation or amputation; patterns variable and not consistent with interruption of normal developmental sequence. Typically sporadic.
Treatment and Management
 Topical lubricants if mild corneal exposure.
Topical lubricants if mild corneal exposure.
 Surgical closure, direct or with mobilization of adjacent tissue. Large defects may require eyelid reconstruction in early infancy to prevent severe corneal damage.
Surgical closure, direct or with mobilization of adjacent tissue. Large defects may require eyelid reconstruction in early infancy to prevent severe corneal damage.
Differential Diagnosis
 Not likely to be mistaken for lid coloboma, but sometimes having similar implications, is exorbitism, the exophthalmic condition of eyes with very shallow bony orbits due to maldevelopment. Seen mainly in association with craniosynostosis (premature closure of cranial sutures) involving midfacial hypoplasia and oxycephaly (tower skull).
Not likely to be mistaken for lid coloboma, but sometimes having similar implications, is exorbitism, the exophthalmic condition of eyes with very shallow bony orbits due to maldevelopment. Seen mainly in association with craniosynostosis (premature closure of cranial sutures) involving midfacial hypoplasia and oxycephaly (tower skull).
 Principal forms are Crouzon syndrome, Apert syndrome (Crouzon plus syndactyly), and Pfeiffer syndrome (Crouzon plus broad thumbs).
Principal forms are Crouzon syndrome, Apert syndrome (Crouzon plus syndactyly), and Pfeiffer syndrome (Crouzon plus broad thumbs).
 Caused by allelic mutations of the gene FGFR2, chromosome locus 10q26, with autosomal dominant transmission.
Caused by allelic mutations of the gene FGFR2, chromosome locus 10q26, with autosomal dominant transmission.
 In severe cases with markedly inadequate eyelid protection, serious corneal damage may occur in a short time, occasionally necessitating surgical reconstruction in the newborn period.
In severe cases with markedly inadequate eyelid protection, serious corneal damage may occur in a short time, occasionally necessitating surgical reconstruction in the newborn period.
 Other concerns include increased intracranial pressure leading to papilledema and optic atrophy (p. 472), characteristic V-pattern strabismus (p. 501), lacrimal drainage obstruction in some cases (p. 429), and varying degrees of developmental delay.
Other concerns include increased intracranial pressure leading to papilledema and optic atrophy (p. 472), characteristic V-pattern strabismus (p. 501), lacrimal drainage obstruction in some cases (p. 429), and varying degrees of developmental delay.
Congenital Ptosis
Summary
Ptosis (bepharoptosis) is unilateral or bilateral drooping of the upper eyelid. Congenital “dystrophic” ptosis, resulting from isolated maldevelopment of the levator palpebrae muscle (muscle tissue replaced by fibrous connective tissue), is by far the most common cause of significant lid droop in childhood.
Demographics
 Usually unilateral but bilateral (symmetric or asymmetric) not uncommon.
Usually unilateral but bilateral (symmetric or asymmetric) not uncommon.
 Usually sporadic, but familial occurrence of both unilateral and bilateral congenital ptosis is sometimes seen.
Usually sporadic, but familial occurrence of both unilateral and bilateral congenital ptosis is sometimes seen.
Symptoms and Signs
 Ptosis in primary gaze position from birth; palpebral fissure height typically decreased 3 to 4 mm; may improve somewhat during early infancy.
Ptosis in primary gaze position from birth; palpebral fissure height typically decreased 3 to 4 mm; may improve somewhat during early infancy.
 Reduced levator palpebrae function and lid excursion, resulting in relative upper lid elevation in downgaze, sometimes to a level higher than that of the opposite normal eye in unilateral cases. This motivates a large majority of children with significant congenital ptosis to adopt a compensatory chin-elevated head posture, which usually permits maintenance of normal visual function.
Reduced levator palpebrae function and lid excursion, resulting in relative upper lid elevation in downgaze, sometimes to a level higher than that of the opposite normal eye in unilateral cases. This motivates a large majority of children with significant congenital ptosis to adopt a compensatory chin-elevated head posture, which usually permits maintenance of normal visual function.
 Use of frontalis contraction to raise lid level.
Use of frontalis contraction to raise lid level.
 Anisometropia, typically astigmatic, may develop.
Anisometropia, typically astigmatic, may develop.
 Amblyopia seldom occurs in the absence of constant strabismus or severe anisometropia. When a child with significant congenital ptosis fails to use chin-up posture, concern about vision is much increased, and there may be need for early surgical intervention.
Amblyopia seldom occurs in the absence of constant strabismus or severe anisometropia. When a child with significant congenital ptosis fails to use chin-up posture, concern about vision is much increased, and there may be need for early surgical intervention.
Table 8.2 Less common causes of childhood ptosis
Treatment and Management
 When vision is not threatened (i.e., is protected by use of chin-up head posture), surgical repair is usually best delayed until age 3 to 5 years.
When vision is not threatened (i.e., is protected by use of chin-up head posture), surgical repair is usually best delayed until age 3 to 5 years.
 With fair to good levator function, and in most unilateral cases, levator resection is appropriate, usually in large amount (15 to 20 mm) via anterior (skin) incision.
With fair to good levator function, and in most unilateral cases, levator resection is appropriate, usually in large amount (15 to 20 mm) via anterior (skin) incision.
 When levator function is poor, and in most bilateral cases, frontalis suspension is the preferred surgical treatment, preferably done with autogenous fascia.
When levator function is poor, and in most bilateral cases, frontalis suspension is the preferred surgical treatment, preferably done with autogenous fascia.
Differential Diagnosis
Many dysmorphic syndromes may include ptosis. Some conditions in which ptosis is acquired and/or associated with other significant ocular or systemic findings are summarized in Table 8.2 (Figure 8.3).
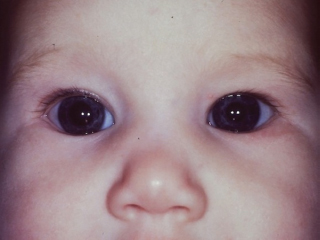
FIGURE 8.3. Congenital Horner syndrome. Note smaller size of left palpebral fissure due to displacement of both upper and lower lid margins, and relative miosis of left pupil. Heterochromia is not evident in this blue-eyed baby.
Eyelid Tic
Summary
Tic is a purposeless repetitive movement that can be (temporarily) voluntarily suppressed. Eyelid blinking is the most common form of tic.
Demographics
 More than 10% of children are affected at some point, mostly boys.
More than 10% of children are affected at some point, mostly boys.
 Typical onset 6 to 10 years old, sometimes earlier.
Typical onset 6 to 10 years old, sometimes earlier.
Symptoms and Signs
 Frequent forceful eyelid closure, usually bilateral but sometimes unilateral winking.
Frequent forceful eyelid closure, usually bilateral but sometimes unilateral winking.
 Eye rotation tics, typically forceful oblique conjugate gaze movements, may be seen in addition or alternatively.
Eye rotation tics, typically forceful oblique conjugate gaze movements, may be seen in addition or alternatively.
 Ocular surface pathology should be ruled out, especially foreign body of the cornea or tarsal conjunctiva, but normal examination is the rule.
Ocular surface pathology should be ruled out, especially foreign body of the cornea or tarsal conjunctiva, but normal examination is the rule.
Treatment and Management
 Resolves spontaneously within a few months in the great majority of cases. A small minority of affected children may develop chronic tic disorder (lasting more than 12 months) or Tourette syndrome (multiple chronic motor and vocal tics).
Resolves spontaneously within a few months in the great majority of cases. A small minority of affected children may develop chronic tic disorder (lasting more than 12 months) or Tourette syndrome (multiple chronic motor and vocal tics).
 May be aggravated by stress but not caused by or typically associated with psychopathology. Psychiatric referral is rarely indicated.
May be aggravated by stress but not caused by or typically associated with psychopathology. Psychiatric referral is rarely indicated.
Deficient Tear Production in Childhood
Dry eye in the pediatric population is far from the ubiquitous problem seen in adults. When it does occur, there is often little or no need for treatment, fortunately for the parents of affected infants and young children who may be more bothered by the instillation of tear supplement drops than by the condition itself. Corneal damage is most likely when there is associated lack of sensation, or when the quality as well as quantity of tear production is affected. A number of disorders in this category are summarized in Table 8.3.
Nasolacrimal Duct Obstruction
Summary
Complete congenital obstruction of the nasolacrimal duct (NLD) commonly results from membranous occlusion, most often at the lower end of the structure where it enters the nose beneath the inferior turbinate (valve of Hasner). Partial obstruction or stenosis may be caused by narrowing of the lacrimal drainage passage at any point below the sac.
Table 8.3. Causes of dry eye in infants and children
Demographics
 Complete obstruction occurs in up to 10% of all newborns.
Complete obstruction occurs in up to 10% of all newborns.
 Initially bilateral in one third of cases.
Initially bilateral in one third of cases.
Symptoms and Signs
 Complete obstruction: constant epiphora and mucus accumulation; severity may be quite variable from patient to patient and from time to time in an individual. Lashes typically bind together in “fascicles.”
Complete obstruction: constant epiphora and mucus accumulation; severity may be quite variable from patient to patient and from time to time in an individual. Lashes typically bind together in “fascicles.”
 Partial obstruction: intermittent epiphora, mainly at times of increased tear production; and intermittent mucus accumulation, typically associated with upper respiratory infection or congestion (which presumably results in temporary complete blockage of the stenotic passage by swollen mucous membranes.)
Partial obstruction: intermittent epiphora, mainly at times of increased tear production; and intermittent mucus accumulation, typically associated with upper respiratory infection or congestion (which presumably results in temporary complete blockage of the stenotic passage by swollen mucous membranes.)
 Mucopurulent reflux on massage of lacrimal sac sometimes present with complete obstruction, not with partial.
Mucopurulent reflux on massage of lacrimal sac sometimes present with complete obstruction, not with partial.
 Mild redness and maceration of lower eyelid skin may result from chronic wetness and rubbing, but conjunctival injection is seldom seen.
Mild redness and maceration of lower eyelid skin may result from chronic wetness and rubbing, but conjunctival injection is seldom seen.
 Acute dacryocystitis and periorbital cellulitis, with marked redness and swelling of eyelid skin (localized and generalized, respectively), are uncommon complications (Figure 8.4; see also discussion of lacrimal sac mucocele below.)
Acute dacryocystitis and periorbital cellulitis, with marked redness and swelling of eyelid skin (localized and generalized, respectively), are uncommon complications (Figure 8.4; see also discussion of lacrimal sac mucocele below.)
Special Tests
 Culture of discharge is not routinely indicated. Gram-positive isolates are obtainable from most specimens, but Gram-negatives and multiple flora are also common; treatment is seldom influenced by test results.
Culture of discharge is not routinely indicated. Gram-positive isolates are obtainable from most specimens, but Gram-negatives and multiple flora are also common; treatment is seldom influenced by test results.
 Dye disappearance test: Observe eye with blue light following instillation of 1% fluorescein solution. Persistence of dye after 5 minutes is abnormal. Fluorescein may or may not become visible in the nose when passage is patent.
Dye disappearance test: Observe eye with blue light following instillation of 1% fluorescein solution. Persistence of dye after 5 minutes is abnormal. Fluorescein may or may not become visible in the nose when passage is patent.
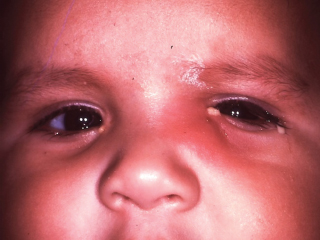
FIGURE 8.4. Acute dacryocystitis in an older infant with congenital NLD obstruction. More typically, this situation develops in a newborn with lacrimal sac mucocele.
Disease Course
 Ninety percent resolve spontaneously by age 12 months, usually with abrupt improvement in symptoms as membranous obstruction disintegrates.
Ninety percent resolve spontaneously by age 12 months, usually with abrupt improvement in symptoms as membranous obstruction disintegrates.
 Symptoms of partial obstruction may persist after resolution of complete obstruction; resolve gradually (months to years) as facial growth increases NLD size and capacity.
Symptoms of partial obstruction may persist after resolution of complete obstruction; resolve gradually (months to years) as facial growth increases NLD size and capacity.
Treatment and Management
 Lacrimal sac massage: Apply firm pressure with fingertip initially over medial canthus, and then slide downward while maintaining pressure. Several times per day; with each diaper change is a convenient routine.
Lacrimal sac massage: Apply firm pressure with fingertip initially over medial canthus, and then slide downward while maintaining pressure. Several times per day; with each diaper change is a convenient routine.
 Topical antibiotic: Drops preferred (unlike ointment, enter sac, and do not blur vision); polymixin-trimethoprim (Polytrim) a good choice. Use chronically as necessary (one to four times per day) to minimize mucopurulent discharge.
Topical antibiotic: Drops preferred (unlike ointment, enter sac, and do not blur vision); polymixin-trimethoprim (Polytrim) a good choice. Use chronically as necessary (one to four times per day) to minimize mucopurulent discharge.
 Use of topical corticosteroid is suggested by some authorities, but in this author’s opinion is never necessary or justified, in view of multiple risks to the infant from chronic steroid exposure including infection (highly septic ocular environment with NLD obstruction, despite concurrent antibiotic administration), intraocular pressure (IOP) elevation (steroid responders more frequent at younger ages and IOP difficult to monitor), and adrenocortical suppression (low body mass compared to dosage).
Use of topical corticosteroid is suggested by some authorities, but in this author’s opinion is never necessary or justified, in view of multiple risks to the infant from chronic steroid exposure including infection (highly septic ocular environment with NLD obstruction, despite concurrent antibiotic administration), intraocular pressure (IOP) elevation (steroid responders more frequent at younger ages and IOP difficult to monitor), and adrenocortical suppression (low body mass compared to dosage).
 Probing in operating room with general anesthesia if symptoms of complete obstruction persist beyond age 12 months. Earlier probing (usually after age 3 months, in outpatient setting with infant awake and securely wrapped, topical anesthesia) when symptoms are particularly bothersome, or preference to avoid general anesthesia. If initial probing is unsuccessful, repeat under general anesthesia.
Probing in operating room with general anesthesia if symptoms of complete obstruction persist beyond age 12 months. Earlier probing (usually after age 3 months, in outpatient setting with infant awake and securely wrapped, topical anesthesia) when symptoms are particularly bothersome, or preference to avoid general anesthesia. If initial probing is unsuccessful, repeat under general anesthesia.
 Simple probing is generally not effective for partial obstruction. If symptoms remain seriously bothersome after age 1 to 2 years, consider balloon catheter dilatation or silicone intubation under general anesthesia. A similar surgical approach is appropriate for persistent symptoms after repeated probing for complete obstruction. Dacryocystorhinostomy is rarely required for congenital NLD obstruction.
Simple probing is generally not effective for partial obstruction. If symptoms remain seriously bothersome after age 1 to 2 years, consider balloon catheter dilatation or silicone intubation under general anesthesia. A similar surgical approach is appropriate for persistent symptoms after repeated probing for complete obstruction. Dacryocystorhinostomy is rarely required for congenital NLD obstruction.
Differential Diagnosis
 In punctal atresia, impatency of the upper lacrimal drainage system may result from occlusion of the punctual opening by a thin membrane, absence of punctum and papilla, or partial or complete failure of canalicular differentiation. The lower system (sac and NLD) is generally intact.
In punctal atresia, impatency of the upper lacrimal drainage system may result from occlusion of the punctual opening by a thin membrane, absence of punctum and papilla, or partial or complete failure of canalicular differentiation. The lower system (sac and NLD) is generally intact.
Epiphora is present, but no sticky mucoid discharge; symptoms are generally much less bothersome than with NLD obstruction, because there is no contribution from sac-secreted mucus or proliferation of bacteria in protein-rich sac fluid.
If location of punctum can be identified visually, it is often possible to cure the condition simply by penetrating the membrane with a pointed dilator, needle, or narrow blade.
Absent punctual differentiation, it is sometimes still possible to surgically establish access to the canaliculus; maintaining patency requires silicone tube stenting for an extended time (months). In severe cases with bothersome symptoms, there may be indication for conjunctivodacryocystorhinostomy (Jones tube), though generally not until later childhood.
 Lacrimal drainage insufficiency from congenital maldevelopment or acquired scarring is seen in association with a variety of disorders, including Down syndrome (p. 425), Cornelia de Lange syndrome (p. 426), Crouzon syndrome and related craniosynostoses (p. 427), ectodermal dysplasia (Table 8.3), Stevens-Johnson syndrome (p. 435), and Rubinstein-Taybi syndrome (p. 444). In some such cases, tearing may be aggravated by accompanying ocular surface or lid margin problems (e.g., blepharitis in Down and de Lange syndromes, ectodermal dysplasia; trichiasis or keratitis in Stevens-Johnson), while in others, it may be diminished due to decreased tear production (Table 8.3).
Lacrimal drainage insufficiency from congenital maldevelopment or acquired scarring is seen in association with a variety of disorders, including Down syndrome (p. 425), Cornelia de Lange syndrome (p. 426), Crouzon syndrome and related craniosynostoses (p. 427), ectodermal dysplasia (Table 8.3), Stevens-Johnson syndrome (p. 435), and Rubinstein-Taybi syndrome (p. 444). In some such cases, tearing may be aggravated by accompanying ocular surface or lid margin problems (e.g., blepharitis in Down and de Lange syndromes, ectodermal dysplasia; trichiasis or keratitis in Stevens-Johnson), while in others, it may be diminished due to decreased tear production (Table 8.3).
 Epiphora may be a symptom of congenital glaucoma. Unlike NLD obstruction, tearing is associated with clear nasal discharge, as well as corneal enlargement and photophobia in most cases.
Epiphora may be a symptom of congenital glaucoma. Unlike NLD obstruction, tearing is associated with clear nasal discharge, as well as corneal enlargement and photophobia in most cases.
References
Katowitz IA, Welsh MG. Timing of initial probing and irrigation in congenital nasolacrimal duct obstruction. Ophthalmology 1987;94:698–705.
Lueder GT. Balloon catheter dilation for treatment of older children with nasolacrimal duct obstruction. Arch Ophthalmol 2002;120:1685–1688.
Lacrimal Sac Mucocele
Summary
Dacryocystocele (dacryocele, amniotocele, mucocele) occurs when there is obstruction of lacrimal drainage both above and below the sac, leading to distention and frequently development of secondary infection.
Etiology
Lower obstruction is typically membranous; upper may be anatomic or physiologic (i.e., blockage of retrograde passage by unusually competent valve of Rosenmuller). Mucus secretion by goblet cells lining the sac leads to increased volume and pressure.
Symptoms and Signs
 Bluish mass in the medial canthal region, inferior to the canthal tendon, typically about 1 cm diameter and present at or soon after birth.
Bluish mass in the medial canthal region, inferior to the canthal tendon, typically about 1 cm diameter and present at or soon after birth.
 Infection (dacryocystitis) usually develops during the first month of life if the condition persists, with onset of redness and swelling of overlying skin.
Infection (dacryocystitis) usually develops during the first month of life if the condition persists, with onset of redness and swelling of overlying skin.
 Pressure against lower membranous obstruction may create a cystlike outpouching into the nose that can cause respiratory distress.
Pressure against lower membranous obstruction may create a cystlike outpouching into the nose that can cause respiratory distress.
Treatment and Management
 Digital pressure over the distended sac (may need to be fairly vigorous) can sometimes twist open the valve of Rosenmuller and allow decompression via puncta; occasionally, this maneuver may rupture lower membrane and establish drainage into the nose.
Digital pressure over the distended sac (may need to be fairly vigorous) can sometimes twist open the valve of Rosenmuller and allow decompression via puncta; occasionally, this maneuver may rupture lower membrane and establish drainage into the nose.
 Topical antibiotic drop may help prevent development of infection.
Topical antibiotic drop may help prevent development of infection.
 Dacryocystitis requires treatment with systemic as well as topical antibiotic; hospital admission and intravenous administration are generally indicated for a newborn. Appropriate drug is broad spectrum with good coverage for Gram-positive respiratory flora, such as a cephalosporin.
Dacryocystitis requires treatment with systemic as well as topical antibiotic; hospital admission and intravenous administration are generally indicated for a newborn. Appropriate drug is broad spectrum with good coverage for Gram-positive respiratory flora, such as a cephalosporin.
 Probing to establish full patency of drainage system should be done if possible. This is challenging in the newborn due to small size and distorted anatomy; general anesthesia and ENT assistance may be required to deal definitively with lower obstruction. Probing of the upper system to decompress the sac, in combination with appropriate nonsurgical management, is sufficient treatment for the acute situation in most cases.
Probing to establish full patency of drainage system should be done if possible. This is challenging in the newborn due to small size and distorted anatomy; general anesthesia and ENT assistance may be required to deal definitively with lower obstruction. Probing of the upper system to decompress the sac, in combination with appropriate nonsurgical management, is sufficient treatment for the acute situation in most cases.
Differential Diagnosis
 With encephalocele, swelling is generally above the canthal tendon and larger than 1 cm.
With encephalocele, swelling is generally above the canthal tendon and larger than 1 cm.
 Hemangioma is seldom present at birth, or confined to the region of the lacrimal sac.
Hemangioma is seldom present at birth, or confined to the region of the lacrimal sac.
Reference
Paysse EA, Coats DK, Bernstein JM, et al. Management and complications of congenital dacryocele with concurrent intranasal mucocele. J AAPOS 2000;4:46–53.
INFECTIOUS AND IMMUNOLOGIC DISORDERS OF OCULAR SURFACE AND ADNEXA
Neonatal Conjunctivitis (Ophthalmia Neonatorum)
Summary
Ophthalmia neonatorum is purulent conjunctivitis occurring in the first month of life. It is important to diagnose and treat this condition promptly.
Etiology
Conjunctival infection is acquired during passage through the birth canal or perinatally, especially if amniotic membranes rupture early. Timing of onset varies with etiologic agent (Table 8.4).
Symptoms and Signs
 Discharge moderate mucopurulent (bacterial, chlamydial) to hyperpurulent (gonococcus).
Discharge moderate mucopurulent (bacterial, chlamydial) to hyperpurulent (gonococcus).
 Lid edema, mild to marked.
Lid edema, mild to marked.
 Conjunctival hyperemia and swelling, occasionally membranes. Papillary but not follicular reaction (Figure 8.2A).
Conjunctival hyperemia and swelling, occasionally membranes. Papillary but not follicular reaction (Figure 8.2A).
 Corneal involvement suggests Gram-negative bacteria (especially gonococcus or pseudomonas) or herpes simplex virus (HSV).
Corneal involvement suggests Gram-negative bacteria (especially gonococcus or pseudomonas) or herpes simplex virus (HSV).
Systemic Findings
 Chlamydial conjunctivitis is often followed by development of severe pneumonitis during the first 6 weeks of life.
Chlamydial conjunctivitis is often followed by development of severe pneumonitis during the first 6 weeks of life.
 Life-threatening systemic infection with HSV is a serious concern.
Life-threatening systemic infection with HSV is a serious concern.
Table 8.4 Causes of neonatal conjunctivitis
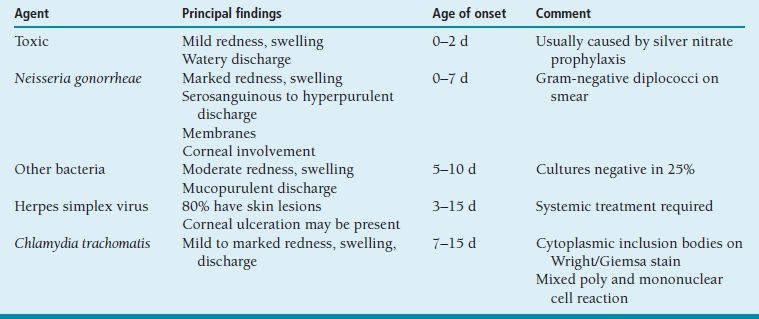
Special Tests
 All newborns with conjunctivitis should have conjunctival discharge Gram stained, and plated on blood, chocolate, and Thayer-Martin media for culture and sensitivity testing.
All newborns with conjunctivitis should have conjunctival discharge Gram stained, and plated on blood, chocolate, and Thayer-Martin media for culture and sensitivity testing.
 Conjunctival scraping (vigorous enough to contain a sufficient quantity of cells) should be inoculated into Chlamydia culture medium (may take up to 7 days to grow out); and may also be evaluated by Wright or Giemsa staining (to look for blue intracytoplasmic inclusion bodies) or immunologic testing (quicker but less reliable diagnostic methods) (Figure 8.5B).
Conjunctival scraping (vigorous enough to contain a sufficient quantity of cells) should be inoculated into Chlamydia culture medium (may take up to 7 days to grow out); and may also be evaluated by Wright or Giemsa staining (to look for blue intracytoplasmic inclusion bodies) or immunologic testing (quicker but less reliable diagnostic methods) (Figure 8.5B).
 Serologic testing for syphilis and human immunodeficiency virus should be obtained in infants with gonococcal or Chlamydia infection.
Serologic testing for syphilis and human immunodeficiency virus should be obtained in infants with gonococcal or Chlamydia infection.
Treatment and Management
 Because of the possibility of penicillin resistance, sight-threatening gonococcal infection is usually treated with a systemic third-generation cephalosporin (ceftriaxone or cefotaxime); topical antibiotics and frequent saline irrigation are often used in addition, but of questionable value.
Because of the possibility of penicillin resistance, sight-threatening gonococcal infection is usually treated with a systemic third-generation cephalosporin (ceftriaxone or cefotaxime); topical antibiotics and frequent saline irrigation are often used in addition, but of questionable value.
 Chlamydia infection is treated with oral erythromycin 40 to 50 mg/kg per day (usually in four divided doses) for 2 to 4 weeks. Parents and sexual partners must be advised and treated for Chlamydia infection and other sexually transmitted diseases.
Chlamydia infection is treated with oral erythromycin 40 to 50 mg/kg per day (usually in four divided doses) for 2 to 4 weeks. Parents and sexual partners must be advised and treated for Chlamydia infection and other sexually transmitted diseases.
 Systemic treatment is also indicated for neonatal ocular HSV infection, typically intravenous acyclovir 30 mg/kg per day (usually in three divided doses) for 2 weeks. Topical trifluridine may or may not be helpful.
Systemic treatment is also indicated for neonatal ocular HSV infection, typically intravenous acyclovir 30 mg/kg per day (usually in three divided doses) for 2 weeks. Topical trifluridine may or may not be helpful.
 Conjunctivitis without corneal involvement caused by other bacteria is treated with erythromycin ointment or bacitracin-polymixin ophthalmic ointment three times daily for 1 to 2 weeks.
Conjunctivitis without corneal involvement caused by other bacteria is treated with erythromycin ointment or bacitracin-polymixin ophthalmic ointment three times daily for 1 to 2 weeks.
 Prophylaxis for ophthalmia neonatorum is mandatory in all American states and Canadian provinces, using erythromycin ointment, tetracycline ointment, or 1% silver nitrate solution. Toxic conjunctivitis caused by silver nitrate is common and resolves spontaneously in several days. In the developing world, 2.5% povidone-iodine (Betadine) has been shown to be a highly effective, safe, and inexpensive method of prophylaxis, which may actually have advantages over methods routinely used in North America.
Prophylaxis for ophthalmia neonatorum is mandatory in all American states and Canadian provinces, using erythromycin ointment, tetracycline ointment, or 1% silver nitrate solution. Toxic conjunctivitis caused by silver nitrate is common and resolves spontaneously in several days. In the developing world, 2.5% povidone-iodine (Betadine) has been shown to be a highly effective, safe, and inexpensive method of prophylaxis, which may actually have advantages over methods routinely used in North America.
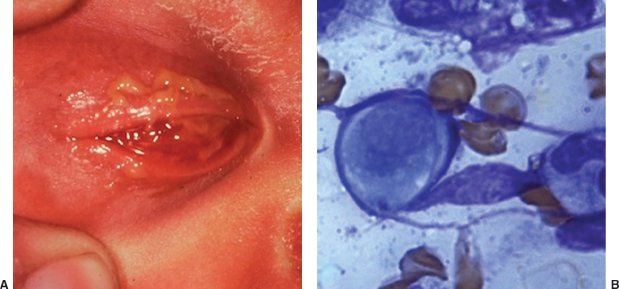
FIGURE 8.5. Chlamydia conjunctivitis in a newborn. A: Severe inflammation of conjunctiva and eyelids. B: Wright-stained scraping showing epithelial cell with blue cytoplasmic inclusion body.
Differential Diagnosis
Acute conjunctivitis with onset beyond age 1 month is assumed to have been acquired postnatally, and in otherwise healthy infants and children is usually caused by Gram-positive respiratory tract flora; occasionally by adenovirus. Treatment with a topical antibiotic drop or ointment that provides good antibacterial coverage, such as polymixin-trimethoprim or erythromycin, can be initiated without need for culture in most cases, unless there is evidence suggestive of HSV infection (e.g., vesicles on the lips or eyelid skin, history of recent HSV outbreak in a close contact, corneal involvement).
References
Isenberg SJ, Apt L, Wood M. A controlled trial of povidone-iodine as prophylaxis against ophthalmia neonatorum. N Engl J Med 1995;332:562–566.
Weiss A, Brinser JH, Stewart VN. Acute conjunctivitis in childhood. J Pediatr 1993;122:10–14.
Preseptal and Orbital Cellulitis
Summary
Preseptal (periorbital) cellulitis is bacterial infection diffusely involving the eyelids. Orbital cellulitis is infection involving tissues posterior to the orbital septum.
Etiology
 Preseptal cellulitis may arise from infection of periocular skin following superficial trauma, primary skin infection (impetigo, herpes simplex), conjunctivitis with invasion of deeper tissues, or extension of upper respiratory infection from paranasal sinuses.
Preseptal cellulitis may arise from infection of periocular skin following superficial trauma, primary skin infection (impetigo, herpes simplex), conjunctivitis with invasion of deeper tissues, or extension of upper respiratory infection from paranasal sinuses.
 By far the most common cause of orbital cellulitis is extension of infection from adjacent sinus cavities, especially the ethomoids. Infection may also arise from deep orbital trauma or bacteremia (see discussion of panophthalmitis below).
By far the most common cause of orbital cellulitis is extension of infection from adjacent sinus cavities, especially the ethomoids. Infection may also arise from deep orbital trauma or bacteremia (see discussion of panophthalmitis below).
 Causative organism in all situations usually Gram-positive cocci, most often Streptococcus sp.
Causative organism in all situations usually Gram-positive cocci, most often Streptococcus sp.
 Haemophilus influenzae was once a common cause of periorbital infection in childhood, presenting with rapid development of marked eyelid swelling and characteristic violaceous skin discoloration, often accompanied by signs of systemic sepsis and life-threatening meningitis. Incidence has dramatically decreased since immunization became routine in the 1980s.
Haemophilus influenzae was once a common cause of periorbital infection in childhood, presenting with rapid development of marked eyelid swelling and characteristic violaceous skin discoloration, often accompanied by signs of systemic sepsis and life-threatening meningitis. Incidence has dramatically decreased since immunization became routine in the 1980s.
 Fungal infection (especially mucormycosis) may cause severe orbital cellulitis with rapid life-threatening progression in diabetic or immunosuppressed patients.
Fungal infection (especially mucormycosis) may cause severe orbital cellulitis with rapid life-threatening progression in diabetic or immunosuppressed patients.
Symptoms and Signs
 Marked diffuse edema and erythema of the upper and the lower eyelids typically develop over 24 to 48 hours in both preseptal and orbital cellulitis. Some edema fluid may track across the nose to cause mild swelling of the contralateral eyelids; this does not generally indicate spread or worsening of infection.
Marked diffuse edema and erythema of the upper and the lower eyelids typically develop over 24 to 48 hours in both preseptal and orbital cellulitis. Some edema fluid may track across the nose to cause mild swelling of the contralateral eyelids; this does not generally indicate spread or worsening of infection.
 Skin lesions (of similar vesicular or crusted appearance) may indicate impetigo or HSV infection.
Skin lesions (of similar vesicular or crusted appearance) may indicate impetigo or HSV infection.
 Conjunctival injection and discharge suggest preseptal infection; mild to severe chemosis is associated with orbital infection.
Conjunctival injection and discharge suggest preseptal infection; mild to severe chemosis is associated with orbital infection.
 Proptosis and limited ocular rotation present in orbital cellulitis, absent in preseptal. May be difficult to detect on examination of uncooperative sick child.
Proptosis and limited ocular rotation present in orbital cellulitis, absent in preseptal. May be difficult to detect on examination of uncooperative sick child.
 Relative afferent papillary defect in orbital cellulitis is the most reliable and important sign of optic nerve compression, indicating serious threat to vision and urgent need for surgical intervention. Pupils should be monitored critically during early days of treatment.
Relative afferent papillary defect in orbital cellulitis is the most reliable and important sign of optic nerve compression, indicating serious threat to vision and urgent need for surgical intervention. Pupils should be monitored critically during early days of treatment.
 Systemic signs of sepsis, including lethargy, fever, and leukocytosis, are sometimes seen, more often with orbital cellulitis than with preseptal. Symptoms of upper respiratory infection or sinus disease (headache, nasal congestion) commonly reported.
Systemic signs of sepsis, including lethargy, fever, and leukocytosis, are sometimes seen, more often with orbital cellulitis than with preseptal. Symptoms of upper respiratory infection or sinus disease (headache, nasal congestion) commonly reported.
Special Tests
 Computed tomography definitively distinguishes preseptal and orbital cellulitis. Typically shows extensive sinusitis in association with orbital cellulitis. May reveal subperiosteal abscess (typically between medial orbital wall and medial rectus muscle), sometimes with air-fluid level; occasionally, intracranial abscess is also present, especially in adolescents.
Computed tomography definitively distinguishes preseptal and orbital cellulitis. Typically shows extensive sinusitis in association with orbital cellulitis. May reveal subperiosteal abscess (typically between medial orbital wall and medial rectus muscle), sometimes with air-fluid level; occasionally, intracranial abscess is also present, especially in adolescents.
 Conjunctiva should be cultured if mucopurulent discharge present. Blood culture, lumbar puncture seldom necessary.
Conjunctiva should be cultured if mucopurulent discharge present. Blood culture, lumbar puncture seldom necessary.
Treatment and Management
 Both preseptal and orbital cellulitis warrant hospitalization and initial treatment with intravenous antibiotics to cover typical respiratory flora plus other organisms as circumstances suggest. Pediatric infectious disease consultation is highly desirable for medical management.
Both preseptal and orbital cellulitis warrant hospitalization and initial treatment with intravenous antibiotics to cover typical respiratory flora plus other organisms as circumstances suggest. Pediatric infectious disease consultation is highly desirable for medical management.
 Clinical response may not be evident until 48 to 72 hours after optimal medical treatment is begun, but most cases of orbital cellulitis in childhood (even when medial subperiosteal abscess is present) resolve without surgery. The first sign of improvement is reduced lid swelling. Proptosis and motility may not recover fully for weeks.
Clinical response may not be evident until 48 to 72 hours after optimal medical treatment is begun, but most cases of orbital cellulitis in childhood (even when medial subperiosteal abscess is present) resolve without surgery. The first sign of improvement is reduced lid swelling. Proptosis and motility may not recover fully for weeks.
 Aggressive treatment of sinusitis is indicated (with ENT consultation), possibly including endoscopic surgical drainage, which also provides the best route for draining most orbital abscesses (typically medial subperiosteal in location) that do not respond adequately to medical treatment.
Aggressive treatment of sinusitis is indicated (with ENT consultation), possibly including endoscopic surgical drainage, which also provides the best route for draining most orbital abscesses (typically medial subperiosteal in location) that do not respond adequately to medical treatment.
 Sinus drainage via skin incision has a high frequency of complications and is now rarely if ever indicated for orbital cellulitis in childhood.
Sinus drainage via skin incision has a high frequency of complications and is now rarely if ever indicated for orbital cellulitis in childhood.
Differential Diagnosis
 Septic embolization to the orbit associated with bacteremia (often but not always from an identifiable remote focus of infection such as endocarditis or meningitis) may cause panophthalmitis, in which orbital cellulitis is accompanied by endopthalmitis. Loss of vision (usually to bare light perception or worse) and of media clarity (frequently with hypopyon) distinguish this condition from other forms of orbital inflammation. The prognosis is dismal, even with aggressive treatment including intraocular antibiotics; often leads to evisceration or enucleation.
Septic embolization to the orbit associated with bacteremia (often but not always from an identifiable remote focus of infection such as endocarditis or meningitis) may cause panophthalmitis, in which orbital cellulitis is accompanied by endopthalmitis. Loss of vision (usually to bare light perception or worse) and of media clarity (frequently with hypopyon) distinguish this condition from other forms of orbital inflammation. The prognosis is dismal, even with aggressive treatment including intraocular antibiotics; often leads to evisceration or enucleation.
 The appearance of orbital cellulitis in childhood may be mimicked by noninfectious conditions including orbital pseudotumor (see below), and a variety of cancers including rhabdomyosarcoma, advanced retinoblastoma, metastatic neuroblastoma, and orbital involvement by leukemia or lymphoma (described in sections below beginning p. 476).
The appearance of orbital cellulitis in childhood may be mimicked by noninfectious conditions including orbital pseudotumor (see below), and a variety of cancers including rhabdomyosarcoma, advanced retinoblastoma, metastatic neuroblastoma, and orbital involvement by leukemia or lymphoma (described in sections below beginning p. 476).
References
Greenwald MJ, Wohl LG, Sell CH. Metastatic bacterial endophthalmitis: a contemporary reappraisal. Surv Ophthalmol 1986;31:81–101.
Weiss A, Friendly D, Eglin K, et al. Bacterial periorbital and orbital cellulitis in childhood. Ophthalmology 1983;90:195–203.
Inflammatory Orbital Pseudotumor
Summary
Inflammatory pseudotumor typically presents with proptosis and periocular inflammation, without evidence of sepsis or localized infection, although mild fever is sometimes present in pediatric cases.
Symptoms and Signs
 Proptosis (axial and/or downward displacement of the globe) is typical; eyelid ptosis is common.
Proptosis (axial and/or downward displacement of the globe) is typical; eyelid ptosis is common.
 Pain, especially associated with eye movement.
Pain, especially associated with eye movement.
 Bilateral involvement, often asymmetric or sequential, is not uncommon with occurrence in childhood.
Bilateral involvement, often asymmetric or sequential, is not uncommon with occurrence in childhood.
Ophthalmic Findings
 Children often show evidence of anterior uveitis on slit-lamp examination.
Children often show evidence of anterior uveitis on slit-lamp examination.
 Fundus examination may reveal papillitis or exudative retinal detachment.
Fundus examination may reveal papillitis or exudative retinal detachment.
 Strabismus (often with diplopia) and limited ocular rotation when extraocular muscles involved.
Strabismus (often with diplopia) and limited ocular rotation when extraocular muscles involved.
Special Tests
 Orbital imaging with ultrasonography, CT, or MRI may reveal diffuse inflammatory changes, localized swelling of orbital structures including lacrimal gland or extraocular muscles (single or multiple, often involving tendon in contrast to thyroid orbitopathy), or thickening of posterior sclera and choroid.
Orbital imaging with ultrasonography, CT, or MRI may reveal diffuse inflammatory changes, localized swelling of orbital structures including lacrimal gland or extraocular muscles (single or multiple, often involving tendon in contrast to thyroid orbitopathy), or thickening of posterior sclera and choroid.
 Biopsy is seldom necessary, but may be helpful, especially when imaging documents lacrimal gland involvement.
Biopsy is seldom necessary, but may be helpful, especially when imaging documents lacrimal gland involvement.
Treatment and Management
 Initial treatment with high-dose systemic corticosteroid (prednisone 1 mg/kg per day) typically leads to prompt resolution. Recurrence after discontinuation of medication is not unusual, but the disease typically “burns out” eventually after months to years.
Initial treatment with high-dose systemic corticosteroid (prednisone 1 mg/kg per day) typically leads to prompt resolution. Recurrence after discontinuation of medication is not unusual, but the disease typically “burns out” eventually after months to years.
 Although primary orbital lymphoma is very rare in childhood, lack of expected response to steroid treatment should prompt assessment for possible malignancy.
Although primary orbital lymphoma is very rare in childhood, lack of expected response to steroid treatment should prompt assessment for possible malignancy.
Stevens-Johnson Syndrome (Erythema Multiforme)
Summary
Stevens-Johnson Syndrome (SJS) is a severe acute mucocutaneous hypersensitivity disorder that typically involves conjunctiva.
Etiology
The immune response may be triggered by prior viral or other (especially mycoplasma) infection, or by drug exposure (less common in children). Usually, a specific agent cannot be identified.
Symptoms and Signs
 Initially mucopurulent discharge and papillary conjunctival reaction.
Initially mucopurulent discharge and papillary conjunctival reaction.
 In severe cases, extensive membranes develop, causing tenacious adherence of tarsal conjunctiva to ocular surface.
In severe cases, extensive membranes develop, causing tenacious adherence of tarsal conjunctiva to ocular surface.
Systemic Findings
 Preceded by prodromal symptoms of malaise, fever, and upper respiratory or gastrointestinal complaints.
Preceded by prodromal symptoms of malaise, fever, and upper respiratory or gastrointestinal complaints.
 Acute skin involvement is highly variable, ranging from classic discrete round “target” lesions to extensive bullous eruption, with an extreme form known as toxic epidermal necrolysis in which large areas of epidermis slough in response to minor mechanical stress.
Acute skin involvement is highly variable, ranging from classic discrete round “target” lesions to extensive bullous eruption, with an extreme form known as toxic epidermal necrolysis in which large areas of epidermis slough in response to minor mechanical stress.
 Other mucous membrane involvement besides conjunctival may include oral and genital, which are often severe but usually heal without significant scarring.
Other mucous membrane involvement besides conjunctival may include oral and genital, which are often severe but usually heal without significant scarring.
Disease Course
 Acute inflammation subsides in a few weeks.
Acute inflammation subsides in a few weeks.
 Chronic sequelae may range from mild to sight threatening, including tear insufficiency, punctal occlusion, keratinization of conjunctiva, symblepharon (shortening of conjunctival fornices), trichiasis, and cicatricial entropion, with potential for severe secondary corneal damage.
Chronic sequelae may range from mild to sight threatening, including tear insufficiency, punctal occlusion, keratinization of conjunctiva, symblepharon (shortening of conjunctival fornices), trichiasis, and cicatricial entropion, with potential for severe secondary corneal damage.
Treatment and Management
 There is no convincing evidence of benefit from any topical medication, including lubricants, antibiotics, or corticosteroids, and their administration is likely to cause both physical and emotional trauma to the gravely suffering child. Milder cases are best left untreated altogether.
There is no convincing evidence of benefit from any topical medication, including lubricants, antibiotics, or corticosteroids, and their administration is likely to cause both physical and emotional trauma to the gravely suffering child. Milder cases are best left untreated altogether.
 In severe cases, daily separation of fused eyelids is indicated. Use of Desmarres retractors after topical anesthesia is more practical and effective than the traditionally recommended glass rod. This process is excruciating for both the patient and the doctor.
In severe cases, daily separation of fused eyelids is indicated. Use of Desmarres retractors after topical anesthesia is more practical and effective than the traditionally recommended glass rod. This process is excruciating for both the patient and the doctor.
 A recently developed device (ProKera) that combines a symblepharon ring with preserved amniotic membrane shows promise as a means of protecting the cornea and limiting conjunctival adhesion formation in SJS.
A recently developed device (ProKera) that combines a symblepharon ring with preserved amniotic membrane shows promise as a means of protecting the cornea and limiting conjunctival adhesion formation in SJS.
 Postacute phase cicatricial changes require aggressive monitoring and management by experienced clinicians to prevent or minimize corneal damage.
Postacute phase cicatricial changes require aggressive monitoring and management by experienced clinicians to prevent or minimize corneal damage.
Reference
Prendiville JS, Hebert AA, Greenwald MJ, et al. Management of Stevens-Johnson syndrome and toxic epidermal necrolysis in children. J Pediatr 1989;115:881–887.
Vernal Conjunctivitis
Summary
Vernal conjunctivitis is a chronic allergic disorder characterized by seasonal exacerbations.
Demographics
 More common in boys than girls.
More common in boys than girls.
 Frequently a history of asthma, other seasonal allergic manifestations.
Frequently a history of asthma, other seasonal allergic manifestations.
Etiology
Immune mediated by pollen- or mold-specific immunoglobulins E and G (IgE and IgG). Symptoms typically worse in spring and/or fall but may persist year round.
Symptoms and Signs
 Itching always present, proportionate to overall severity.
Itching always present, proportionate to overall severity.
 Redness, tearing, and photophobia are common.
Redness, tearing, and photophobia are common.
 Conjunctival injection, boggy edema; sometimes ropy mucoid discharge.
Conjunctival injection, boggy edema; sometimes ropy mucoid discharge.
 Large “cobblestone” papillae of upper tarsal conjunctiva in palpebral form.
Large “cobblestone” papillae of upper tarsal conjunctiva in palpebral form.
 Limbal vernal disease is characterized by nodular limbal swelling, usually superior sector, often accompanied by small whitish Trantas dots (Figure 8.6).
Limbal vernal disease is characterized by nodular limbal swelling, usually superior sector, often accompanied by small whitish Trantas dots (Figure 8.6).
 Corneal involvement ranges from mild superficial punctate keratitis (mainly superior) to “shield” plaque or ulcer.
Corneal involvement ranges from mild superficial punctate keratitis (mainly superior) to “shield” plaque or ulcer.
Treatment and Management
 Systemic allergy treatment, avoidance of allergens, cold compresses.
Systemic allergy treatment, avoidance of allergens, cold compresses.
 In milder cases, symptomatic relief may be provided by topical tear supplement, decongestant, or antihistamine drops.
In milder cases, symptomatic relief may be provided by topical tear supplement, decongestant, or antihistamine drops.
 Most patients require a mast-cell stabilizer such as cromolyn sodium, lodoxamide (Alomide), or olopatadine (Patanol) during active periods.
Most patients require a mast-cell stabilizer such as cromolyn sodium, lodoxamide (Alomide), or olopatadine (Patanol) during active periods.
 Topical corticosteroid is often necessary, especially to achieve initial control. Use a mild formulation such as loteprednol (Lotemax) or fluorometholone (FML); monitor IOP if used for more than a few weeks.
Topical corticosteroid is often necessary, especially to achieve initial control. Use a mild formulation such as loteprednol (Lotemax) or fluorometholone (FML); monitor IOP if used for more than a few weeks.
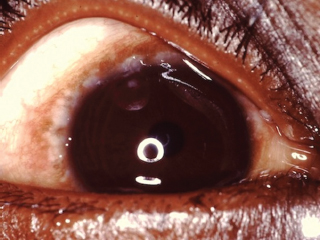
FIGURE 8.6. Limbal vernal conjunctivitis. Note limbal follicles and tiny white Trantas dots.
Differential Diagnosis
Other less common causes of chronic conjunctivitis in childhood should be kept in mind, including molluscum contagiosum, staphylococcal lid margin disease, acne rosacea, ligneous conjunctivitis, and the various dry eye syndromes (Table 8.3).
References
Erzurum SA, Feder RS, Greenwald MJ. Acne rosacea with keratitis in childhood. Arch Ophthalmol 1993;111:228–230.
Friedlaender MH. Conjunctivitis of allergic origin: clinical presentation and differential diagnosis. Surv Ophthalmol 1993;38:105–114.
Hypersensitivity Keratoconjunctivitis (Phlyctenulosis)
Summary
Phlyctenular disease is a localized immunologic disorder of conjunctiva and cornea.
Etiology
The specific cause of hypersensitivity reaction is usually not evident in children. Staphylococcal blepharitis may be the source, but clinical signs are frequently absent. Historical association with tuberculosis and malnutrition seldom applicable in present-day cases.
Symptoms and Signs
 Localized conjunctival injection, unilateral or bilateral.
Localized conjunctival injection, unilateral or bilateral.
 Tearing, photophobia are common; may be severe.
Tearing, photophobia are common; may be severe.
 Slightly elevated, gray to pink, nodular or wedge-shaped limbal lesion, often associated with a small peripheral or paracentral corneal ulcer connected to the limbus by a narrow leash of vessels (Figure 8.7).
Slightly elevated, gray to pink, nodular or wedge-shaped limbal lesion, often associated with a small peripheral or paracentral corneal ulcer connected to the limbus by a narrow leash of vessels (Figure 8.7).
 Significant anterior corneal stromal scarring may result from repeated exacerbations. Rarely significant localized thinning and perforation may occur.
Significant anterior corneal stromal scarring may result from repeated exacerbations. Rarely significant localized thinning and perforation may occur.
Treatment and Management
 Some cases respond well to treatment for blepharitis with lid hygiene and antibiotic application.
Some cases respond well to treatment for blepharitis with lid hygiene and antibiotic application.
 Topical corticosteroid is usually necessary, typically bringing prompt relief of symptoms. Should be tapered as quickly as possible, though recurrence is common.
Topical corticosteroid is usually necessary, typically bringing prompt relief of symptoms. Should be tapered as quickly as possible, though recurrence is common.
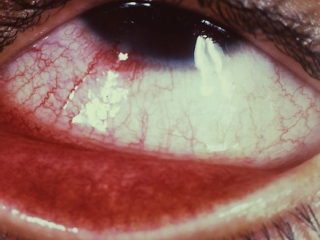
FIGURE 8.7. Phlyctenular keratoconjunctivitis, with lesion at the limbus.
Differential Diagnosis
 Marginal ulcer of the cornea probably represents a variant expression of the same disease process that causes phlyctenular disease. It is much less commonly seen in children than the phlyctenular variety.
Marginal ulcer of the cornea probably represents a variant expression of the same disease process that causes phlyctenular disease. It is much less commonly seen in children than the phlyctenular variety.
 Interstitial keratitis, one of the characteristic late manifestations of congenital syphilis, is now rare in developed countries. Sequential onset of bilateral corneal opacities and edema with deep stromal vascularization occurs abruptly in late childhood or adolescence, usually accompanied by redness and intense photophobia, often with anterior uveitis. Associated findings may include changes in secondary dentition (“Hutchinson teeth”) and progressive hearing loss; pigmentary retinopathy (p. 451).
Interstitial keratitis, one of the characteristic late manifestations of congenital syphilis, is now rare in developed countries. Sequential onset of bilateral corneal opacities and edema with deep stromal vascularization occurs abruptly in late childhood or adolescence, usually accompanied by redness and intense photophobia, often with anterior uveitis. Associated findings may include changes in secondary dentition (“Hutchinson teeth”) and progressive hearing loss; pigmentary retinopathy (p. 451).
Chalazion
Summary
Chalazia (properly pronounced with initial “k” sound) are nodular inflammatory lesions that originate from eyelid meibomian glands. The term internal hordeolum is now used interchangeably with chalazion by most authors.
Demographics
 By far the most common acquired adnexal lesion of childhood, representing a frequent and distressing problem in the pediatric population.
By far the most common acquired adnexal lesion of childhood, representing a frequent and distressing problem in the pediatric population.
 Seen in children as young as 1 year old, both sexes, all ethnic groups.
Seen in children as young as 1 year old, both sexes, all ethnic groups.
Pathology
The material found in a chalazion (for which the name glorp has been suggested) has a very uniform and distinctive gross appearance, consisting of tiny yellow jellylike lobules. Microscopic examination shows granulomatous reaction (including giant cells, epithelioid macrophages, lymphocytes, and plasma cells) surrounding small fat globules.
Symptoms and Signs
 Acute or subacute, mild to moderate localized swelling with redness of overlying skin in the upper or lower eyelid may be the initial manifestation of meibomian gland inflammation. Discomfort is absent to moderate.
Acute or subacute, mild to moderate localized swelling with redness of overlying skin in the upper or lower eyelid may be the initial manifestation of meibomian gland inflammation. Discomfort is absent to moderate.
 Resolution of inflammatory signs occurs over a few days to a week or two, with evolution into painless nodular thickening in the lid, diameter from about 2 to 12 mm, typically a few millimeters from the margin. Nodule may develop without prior inflammatory signs; inflammation may resolve without nodule formation.
Resolution of inflammatory signs occurs over a few days to a week or two, with evolution into painless nodular thickening in the lid, diameter from about 2 to 12 mm, typically a few millimeters from the margin. Nodule may develop without prior inflammatory signs; inflammation may resolve without nodule formation.
Initially skin intact and mobile over nodule; may remain so indefinitely, with stable, gradually increasing, or fluctuating volume.
In some cases after weeks to months, overlying skin becomes thin, red, and scaly, typically accompanied or followed by spontaneous partial external drainage of small amounts of serosanguinous fluid, pus, or glorp. Skin crusting and induration develop as the volume of the lesion diminishes; complete resolution typically long in coming.
Occasionally, a small chalazion may involve only a meibomian orifice and immediately adjacent tissue.
 Localized redness of underlying conjunctiva is typical. Fleshy polypoid excrescense of tissue from conjunctiva may appear in late stages, sometimes containing glorp but usually representing a secondary reactive phenomenon (pyogenic granuloma). These lesions may be shaved off at the (usually narrow) base followed by light cautery; care should be taken to remove also any glorp (often small in quantity) that is still present within underlying tarsus (see discussion of surgical treatment below).
Localized redness of underlying conjunctiva is typical. Fleshy polypoid excrescense of tissue from conjunctiva may appear in late stages, sometimes containing glorp but usually representing a secondary reactive phenomenon (pyogenic granuloma). These lesions may be shaved off at the (usually narrow) base followed by light cautery; care should be taken to remove also any glorp (often small in quantity) that is still present within underlying tarsus (see discussion of surgical treatment below).
 Symptomatic generalized blepharitis or meibomitis present in a minority of cases.
Symptomatic generalized blepharitis or meibomitis present in a minority of cases.
 Vision usually unaffected, but pressure on the cornea may produce deformation and astigmatism in some cases, which reverts to normal once the lesion is gone.
Vision usually unaffected, but pressure on the cornea may produce deformation and astigmatism in some cases, which reverts to normal once the lesion is gone.
Disease Course
 Complete spontaneous resolution may occur within weeks, sometimes very quickly following the acute inflammatory phase, but chronic lesions may persist for many months or even years with stable or fluctuating symptoms and signs.
Complete spontaneous resolution may occur within weeks, sometimes very quickly following the acute inflammatory phase, but chronic lesions may persist for many months or even years with stable or fluctuating symptoms and signs.
 Typically a solitary lesion, but clusters involving one or multiple lids simultaneously or sequentially (usually within a time frame of a few months to a few years) are not unusual in childhood.
Typically a solitary lesion, but clusters involving one or multiple lids simultaneously or sequentially (usually within a time frame of a few months to a few years) are not unusual in childhood.
Treatment and Management
 Warm compresses are the mainstay of conservative management. For maximum effectiveness should be applied for 10 to 15 minutes two to three times per day. Wrapping a moist washcloth around an object that will stay warm for sufficient time after heating (e.g., a microwaved potato or boiled egg) is recommended. Benefit should be evident within 1 to 2 weeks; often no effect on chronic lesions.
Warm compresses are the mainstay of conservative management. For maximum effectiveness should be applied for 10 to 15 minutes two to three times per day. Wrapping a moist washcloth around an object that will stay warm for sufficient time after heating (e.g., a microwaved potato or boiled egg) is recommended. Benefit should be evident within 1 to 2 weeks; often no effect on chronic lesions.
If present, blepharitis or meibomitis should be treated with baby shampoo lid scrubs and topical antibiotic ointment application at bedtime, which may help prevent recurrent chalazia.
Topical and systemic antibiotics and topical corticosteroid are generally ineffective for the chalazion itself.
 Surgery is nearly always curative (if glorp is removed completely) but does not prevent new lesions from developing.
Surgery is nearly always curative (if glorp is removed completely) but does not prevent new lesions from developing.
Sedation (plus local anesthetic infiltration) or general anesthesia is needed for nearly all pediatric patients, including most teenagers.
Make 4- to 5-mm incision with No. 11 or No. 15 blade: vertical through conjunctiva and tarsus if skin intact (taking care not to extend to the lid margin), or horizontal through center of abnormal skin region.
Remove all glorp by scraping with a curette (force directed laterally/medially from center in plane of lid) and scrubbing with cotton-tip swabs. Excise degenerated tissue around skin incision to leave a healthy margin. It is not necessary to remove secretory membrane (“capsule” or “cyst wall”) surrounding the glorp-filled cavity.
No sutures; rapid contraction and healing of external or internal wound by secondary intention with little or no scarring is the rule in children.
Differential Diagnosis
 An external hordeolum or stye originates from an eyelid sebaceous gland of Zeis or apocrine gland of Moll, usually closer to the lid margin and the skin surface (involving the lash line) compared with a chalazion. Inflammatory symptoms and signs are typically greater and more localized for a stye during acute phase; spontaneous resolution occurs in days to a week or two in most cases, sometimes accompanied by drainage of a small amount of pus (no glorp). Medical or surgical treatment is seldom necessary; warm compresses may be helpful.
An external hordeolum or stye originates from an eyelid sebaceous gland of Zeis or apocrine gland of Moll, usually closer to the lid margin and the skin surface (involving the lash line) compared with a chalazion. Inflammatory symptoms and signs are typically greater and more localized for a stye during acute phase; spontaneous resolution occurs in days to a week or two in most cases, sometimes accompanied by drainage of a small amount of pus (no glorp). Medical or surgical treatment is seldom necessary; warm compresses may be helpful.
 Eyelid abscess is the result of suppurative infection, usually caused by Gram-positive cocci, confined to a single eyelid. Usually, the entire eyelid is involved from the time of onset until spontaneous or surgical (via horizontal skin incision) drainage. The relatively large cavity should be packed postdrainage with iodoform gauze strip, which is gradually removed over several days.
Eyelid abscess is the result of suppurative infection, usually caused by Gram-positive cocci, confined to a single eyelid. Usually, the entire eyelid is involved from the time of onset until spontaneous or surgical (via horizontal skin incision) drainage. The relatively large cavity should be packed postdrainage with iodoform gauze strip, which is gradually removed over several days.
 The early acute phase of chalazion can resemble incipient cellulitis or orbital pseudotumor. Insect or spider bites can produce a very similar appearance.
The early acute phase of chalazion can resemble incipient cellulitis or orbital pseudotumor. Insect or spider bites can produce a very similar appearance.
 A variety of less common benign skin lesions may be mistaken for chalazion, including papilloma and pilomatrixoma. Cutaneous malignancy is extremely rare in otherwise healthy children. In questionable cases, and when no typical glorp is recovered, excised tissue should be submitted for pathologic evaluation.
A variety of less common benign skin lesions may be mistaken for chalazion, including papilloma and pilomatrixoma. Cutaneous malignancy is extremely rare in otherwise healthy children. In questionable cases, and when no typical glorp is recovered, excised tissue should be submitted for pathologic evaluation.
 Other nodular subcutaneous lesions including small hemangiomas, dermoid cysts, and rhabdomyosarcoma sometimes have a similar appearance to chalazion (see section on tumors below, beginning p. 474).
Other nodular subcutaneous lesions including small hemangiomas, dermoid cysts, and rhabdomyosarcoma sometimes have a similar appearance to chalazion (see section on tumors below, beginning p. 474).
Table 8.5 Congenital corneal and global abnormalities
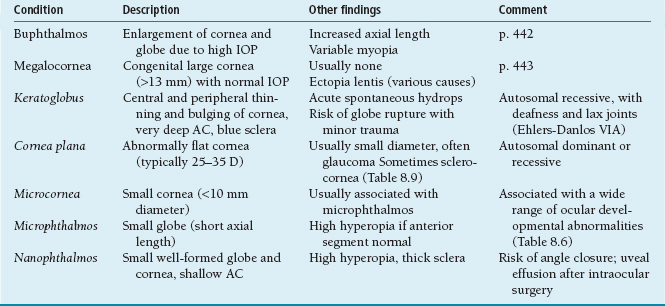
IOP, intraocular pressure; D, diopter; AC, anterior chamber.
Reference
Greenwald MJ. What’s in a chalazion? Surv Ophthalmol 1985; 30:142.
GLOBAL, ANTERIOR SEGMENT, AND LENS DISORDERS
Abnormalities of Corneal Size and Shape
Abnormally large or small size of the cornea or the entire globe, and flatter or steeper than normal corneal curvature, are important indicators of ocular maldevelopment, often but not always having serious implications for eye health and vision (Table 8.5). A wide variety of conditions may result in decreased corneal diameter. (Table 8.6 provides a partial listing.) Early-onset myopia is often found in larger than normal eyes and is also seen in association with a variety of other conditions (Table 8.7).
Keratoconus, the one significant acquired disorder affecting corneal shape, may present in late childhood as unilateral or bilateral progressive myopia with irregular astigmatism that precludes correction to normal vision with spectacles. Visualization of a “cone” by viewing the red fundus reflex remotely with direct ophthalmoscopy can help make this diagnosis, which often is missed in early stages, and may be mistaken for amblyopia (p. 491) or functional vision loss (p. 468). Keratoconus is usually sporadic, but is associated with a number of systemic disorders, including atopic dermatitis (Table 8.12), Down syndrome (p. 425), Crouzon syndrome (p. 427), and Marfan syndrome (p. 444).
Table 8.6 Important associations of microphthalmos and microcornea
Table 8.7 Important associations of myopia in young children
References
Greenwald MJ. Refractive abnormalities in childhood. Pediatr Clin North Am 2003;50:197–212.
Weiss AH, Koussef BG, Ross EA, et al. Complex microphthalmos. Arch Ophthalmol 1989;107:1619–1624.
Anterior Segment Dysgenesis
Anterior segment dysgenesis (formerly known as anterior chamber cleavage syndrome) comprises a variety of ocular developmental disorders attributable to abnormal differentiation of mesenchyme-derived tissue, mainly of neural crest origin (Table 8.8). Presence of anomaly can usually be documented by careful hand-light inspection (Figure 8.8). These conditions most often occur as isolated abnormalities, sometimes are accompanied by other ocular disturbances (e.g., glaucoma), and sometimes present in the context of a complex multisystem disorder. Examples of the last category include Alagille syndrome (Table 8.8), Rubinstein-Taybi syndrome (p. 444) and fetal alcohol syndrome (p. 425). Though not generally included in discussion of this group of disorders, aniridia (Table 8.17) shares a number of features with them, including frequent origin from faulty PAX6 gene expression.
Corneal Opacity in Childhood
Localized or generalized lack of corneal transparency, present at birth or acquired during the early years of life, has numerous and disparate causes, which may involve congenital malformation, edema secondary to elevated IOP or inadequate endothelial function, or storage of abnormally accumulated metabolites (Table 8.9). Scarring from immunologic keratitis (p. 437), or secondary to inadequate tear production (Table 8.3), is covered elsewhere.
Table 8.8 Spectrum of anterior segment dysgenesis

DM, Descemet’s membrane.
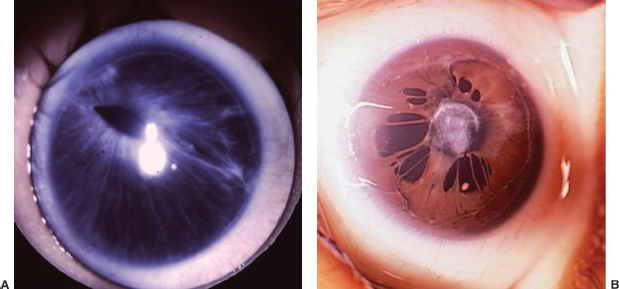
FIGURE 8.8. Anterior segment dysgenesis. A: Rieger anomaly, viewed through a contact goniolens for magnification, with posterior embryotoxon, iridocorneal adhesions, distorted pupil, and iris stromal hypoplasia. B: Complex anomaly with extensive iris adherence to both posterior embryotoxon and relatively large central anterior capsular lens opacity.
Primary Infantile Glaucoma
Summary
Childhood glaucoma most often is present at birth or develops in infancy. Abnormal development of trabecular meshwork structures and consequent elevation of IOP lead to corneal edema and enlargement of the cornea or the entire globe (buphthalmos).
Table 8.9 Causes of corneal clouding or opacification in infants and children
DM, Descemet’s membrane; HSV, herpes simplex virus; MPS, mucopolysaccharidosis.
Demographics
 Sometimes autosomal recessive, especially in Middle Eastern countries.
Sometimes autosomal recessive, especially in Middle Eastern countries.
 Most often sporadic, involving either sex.
Most often sporadic, involving either sex.
Symptoms and Signs
 Corneal enlargement nearly always present, corneal clouding usually present.
Corneal enlargement nearly always present, corneal clouding usually present.
 Epiphora and photophobia are typically present, but frequently absent.
Epiphora and photophobia are typically present, but frequently absent.
Ophthalmic Findings
 Corneal diameter greater than 10 mm in newborn, or greater than 12 mm before age 1 year, is suspicious (Figure 8.9). Any detectable difference in corneal diameter between right and left eyes is potentially significant. Infant corneas larger than parent corneas are cause for concern.
Corneal diameter greater than 10 mm in newborn, or greater than 12 mm before age 1 year, is suspicious (Figure 8.9). Any detectable difference in corneal diameter between right and left eyes is potentially significant. Infant corneas larger than parent corneas are cause for concern.
 Corneal edema, typically stromal and epithelial, usually associated with horizontal or circumferential breaks in Descemet membrane (Haab striae).
Corneal edema, typically stromal and epithelial, usually associated with horizontal or circumferential breaks in Descemet membrane (Haab striae).
 Increased IOP, ranging from low 20s to 40s. Keep in mind that measurements in the awake infant or child, especially those made with TonoPen, may be artifactually elevated, and those made under general anesthesia may be lowered.
Increased IOP, ranging from low 20s to 40s. Keep in mind that measurements in the awake infant or child, especially those made with TonoPen, may be artifactually elevated, and those made under general anesthesia may be lowered.
 Gonioscopic abnormalities (Barkan membrane) classically described but difficult to document.
Gonioscopic abnormalities (Barkan membrane) classically described but difficult to document.
 Myopia ranging from mild to severe. Effect of axial elongation to some extent mitigated by associated corneal and lens flattening.
Myopia ranging from mild to severe. Effect of axial elongation to some extent mitigated by associated corneal and lens flattening.
 Increased optic disc cupping, in most but not all cases reversible after control of IOP.
Increased optic disc cupping, in most but not all cases reversible after control of IOP.
FIGURE 8.9. Infant with glaucoma, right eye. Note enlargement and clouding of cornea.
Treatment and Management
 Estimation of corneal diameter, confirmed as necessary using a ruler (preferably marked with successively increasing multimillimeter intervals), should be a part of every infant eye examination. With practice, it is possible to estimate with accuracy to within 0.5 mm.
Estimation of corneal diameter, confirmed as necessary using a ruler (preferably marked with successively increasing multimillimeter intervals), should be a part of every infant eye examination. With practice, it is possible to estimate with accuracy to within 0.5 mm.
 Examination under anesthesia (EUA) or with monitored sedation is indicated for any infant or young child suspected of having glaucoma. Measure IOP with at least two devices (Schiotz tonometer is quite acceptable), corneal diameter, and if possible axial length using A-scan ultrasonograpy. Perform anterior segment examination with a hand-held slit lamp or operating microscope (including gonioscopy if possible), retinoscopy, and optic nerve examination (with photographic documentation if possible).
Examination under anesthesia (EUA) or with monitored sedation is indicated for any infant or young child suspected of having glaucoma. Measure IOP with at least two devices (Schiotz tonometer is quite acceptable), corneal diameter, and if possible axial length using A-scan ultrasonograpy. Perform anterior segment examination with a hand-held slit lamp or operating microscope (including gonioscopy if possible), retinoscopy, and optic nerve examination (with photographic documentation if possible).
 Initial treatment is often medical, usually with a topical or a systemic carbonic anhydrase inhibitor (CAI) such as acetazolamide (10 to 15 mg/kg per day in two to four divided doses), which may be started before EUA to protect the cornea and the optic nerve from further damage and to improve visualization of angle structures and disc.
Initial treatment is often medical, usually with a topical or a systemic carbonic anhydrase inhibitor (CAI) such as acetazolamide (10 to 15 mg/kg per day in two to four divided doses), which may be started before EUA to protect the cornea and the optic nerve from further damage and to improve visualization of angle structures and disc.
 Definitive treatment in most cases is surgical, beginning if possible with goniotomy or trabeculotomy (angle incision procedures):
Definitive treatment in most cases is surgical, beginning if possible with goniotomy or trabeculotomy (angle incision procedures):
If corneal clarity is adequate, goniotomy (incising trabecular meshwork from within the anterior chamber under direct gonioscopic visualization—ab interno) is preferred by some surgeons.
If the cornea is cloudy, trabeculotomy (tearing through trabecular meshwork with an instrument or suture inserted into Schlemm’s canal through incision under a limbus-based scleral flap—ab externo) is the procedure of choice. Some surgeons prefer trabeculotomy, which more closely resembles other forms of anterior segment microsurgery, for cases with clear cornea as well.
Results from the two procedures have repeatedly been shown to be about the same.
 Inadequate pressure control after two angle incision procedures is indication for trabeculectomy. High failure rates in childhood justify initial adjunctive use of 5-fluorouracil or mitomycin-C.
Inadequate pressure control after two angle incision procedures is indication for trabeculectomy. High failure rates in childhood justify initial adjunctive use of 5-fluorouracil or mitomycin-C.
 Refractory cases may require placement of a tube shunt drainage device, or ciliary body ablation with infrared diode laser or cryotherapy.
Refractory cases may require placement of a tube shunt drainage device, or ciliary body ablation with infrared diode laser or cryotherapy.
 Topical medication may play an extended role in borderline cases, especially after less than desired effect from surgery. Useful agents in addition to CAI include:
Topical medication may play an extended role in borderline cases, especially after less than desired effect from surgery. Useful agents in addition to CAI include:
Beta-blockers, with attention to the possibility of adverse effects in small children with reactive airway disease.
Prostaglandin analog agents, increasingly used for juvenile glaucoma.
Alpha-adrenergic agents, specifically brimonidine (Alphagan), must be used with great caution in children. May cause severe somnolence and life-threatening systemic effects including bradycardia, hypotension, and apnea, especially in children weighing less than 20 kg.
Follow-Up
 Frequent EUAs as necessary. Developmentally normal children over 3 years old can usually be trained to cooperate for applanation tonometry at the slit lamp, after which office follow-up becomes sufficient in most cases.
Frequent EUAs as necessary. Developmentally normal children over 3 years old can usually be trained to cooperate for applanation tonometry at the slit lamp, after which office follow-up becomes sufficient in most cases.
 Treat amblyopia, myopia, and strabismus as necessary.
Treat amblyopia, myopia, and strabismus as necessary.
Differential Diagnosis
 Congenital megalocornea (symmetrical nonprogressive enlargement of clear corneas, with normal IOP and disc appearance) is often but not always familial. An X-linked variety sometimes referred to as anterior megalophthalmos is associated with corneal stromal “mosaic” dystrophy and early adult-onset cataract and glaucoma (Table 8.5). Ectopia lentis from any cause may be associated with increased corneal diameter (p. 444).
Congenital megalocornea (symmetrical nonprogressive enlargement of clear corneas, with normal IOP and disc appearance) is often but not always familial. An X-linked variety sometimes referred to as anterior megalophthalmos is associated with corneal stromal “mosaic” dystrophy and early adult-onset cataract and glaucoma (Table 8.5). Ectopia lentis from any cause may be associated with increased corneal diameter (p. 444).
 Congenital hereditary endothelial dystrophy presents with bilateral corneal clouding without enlargement in infancy. May cause tearing and photophobia. Normal IOP and disc appearance. Often familial: autosomal recessive or dominant (less common, milder) (Table 8.9).
Congenital hereditary endothelial dystrophy presents with bilateral corneal clouding without enlargement in infancy. May cause tearing and photophobia. Normal IOP and disc appearance. Often familial: autosomal recessive or dominant (less common, milder) (Table 8.9).
 Primary juvenile open-angle glaucoma is considerably rarer and more difficult to diagnose than the infantile variety. Corneal size and clarity are typically normal, and the presenting sign is most often progressive myopia, very common for other reasons in the same demographic. Visual field testing is of little value before adolescence. IOP measurements are often artifactually elevated, especially when made using TonoPen. Pressure in the upper teens is higher than average but within normal limits for children, and in the 20s may be tolerated for decades by young eyes. Cup-to-disc ratios of 0.5 or more are not exceptional for normal children, and in fact this finding is far more often physiologic than pathologic in the pediatric population. Family history of glaucoma in adult relatives is rarely significant. Except when there is a well-documented history of glaucoma onset before age 30 in a parent, a child with suspicious discs, progressive myopia, and/or IOP in the 20s should be monitored closely for evidence of progressive optic neuropathy before treatment is initiated.
Primary juvenile open-angle glaucoma is considerably rarer and more difficult to diagnose than the infantile variety. Corneal size and clarity are typically normal, and the presenting sign is most often progressive myopia, very common for other reasons in the same demographic. Visual field testing is of little value before adolescence. IOP measurements are often artifactually elevated, especially when made using TonoPen. Pressure in the upper teens is higher than average but within normal limits for children, and in the 20s may be tolerated for decades by young eyes. Cup-to-disc ratios of 0.5 or more are not exceptional for normal children, and in fact this finding is far more often physiologic than pathologic in the pediatric population. Family history of glaucoma in adult relatives is rarely significant. Except when there is a well-documented history of glaucoma onset before age 30 in a parent, a child with suspicious discs, progressive myopia, and/or IOP in the 20s should be monitored closely for evidence of progressive optic neuropathy before treatment is initiated.
 Rubinstein-Taybi syndrome is a multisystem disorder with dysmorphic features (notably broad thumbs and big toes) that has a wide variety of ophthalmic associations, including glaucoma, cataract, high myopia, and anomalies of the lacrimal drainage system, anterior segment (keratoglobus, dysgenesis), and fundus (coloboma, foveal hypoplasia). Of particular importance is the frequent finding of central disc excavation that may mimic glaucomatous cupping, creating confusion with actual syndrome-related glaucoma, especially when there is associated myopia and corneal abnormality.
Rubinstein-Taybi syndrome is a multisystem disorder with dysmorphic features (notably broad thumbs and big toes) that has a wide variety of ophthalmic associations, including glaucoma, cataract, high myopia, and anomalies of the lacrimal drainage system, anterior segment (keratoglobus, dysgenesis), and fundus (coloboma, foveal hypoplasia). Of particular importance is the frequent finding of central disc excavation that may mimic glaucomatous cupping, creating confusion with actual syndrome-related glaucoma, especially when there is associated myopia and corneal abnormality.
 Infantile glaucoma is an important concern with two of the major neurocutaneous syndromes (p. 486).
Infantile glaucoma is an important concern with two of the major neurocutaneous syndromes (p. 486).
In neurofibromatosis type 1 (NF1), glaucoma is uncommon, nearly always unilateral, and associated with either plexiform neurofibroma of the ipsilateral upper eyelid (which may be a subtle finding in infancy, evident as enlargement of the temporal portion of the lid causing S-shaped margin), or ectropion uveae (eversion of iris pigment epithelial tissue at the pupil margin; rare). Most of these eyes do poorly.
In Sturge-Weber syndrome, glaucoma is common, nearly always associated with ipsilateral eyelid involvement by portwine nevus (usually unilateral, but not infrequently bilateral). Effective treatment is usually possible, but often challenging, in significant part because of the tendency of involved eyes to develop massive choroidal effusion during or after filtering surgery.
 Numerous other ocular and systemic conditions may be associated with glaucoma in childhood. (Table 8.10 provides a partial listing.) Some of these also involve microphthalmos or microcornea (Tables 8.5 and 8.6), in which case corneal diameter and axial length may remain less than normal in the presence of sight-threatening elevation of IOP. Greater than expected growth of a small eye in infancy should raise suspicion of glaucoma.
Numerous other ocular and systemic conditions may be associated with glaucoma in childhood. (Table 8.10 provides a partial listing.) Some of these also involve microphthalmos or microcornea (Tables 8.5 and 8.6), in which case corneal diameter and axial length may remain less than normal in the presence of sight-threatening elevation of IOP. Greater than expected growth of a small eye in infancy should raise suspicion of glaucoma.
Table 8.10 Important associations of glaucoma in childhood
Reference
Beck AD, Freedman S, Kammer J, et al. Aqueous shunt devices compared with trabeculectomy with mitomycin-C for children in the first two years of life. Am J Ophthalmol 2003;136: 994–1000.
Ectopia Lentis (Lens Subluxation or Dislocation)
Summary
Lens displacement (when not attributable to trauma(Figure 8.10A) reflects abnormal laxity of zonular fibers. May occur as an isolated abnormality or in the context of a multisystem disorder.
Ophthalmic Findings
 Displacement of the lens within or perpendicular to its coronal plane. Characteristically superior/posterior in Marfan syndrome (MFS); inferior/anterior in homocystinuria (Figure 8.10B). Regardless of cause, displacement tends to increase during childhood; often undetectable in infancy.
Displacement of the lens within or perpendicular to its coronal plane. Characteristically superior/posterior in Marfan syndrome (MFS); inferior/anterior in homocystinuria (Figure 8.10B). Regardless of cause, displacement tends to increase during childhood; often undetectable in infancy.
 Spherophakia or microspherophakia (increased curvature, decreased diameter), particularly in Weill-Marchesani syndrome.
Spherophakia or microspherophakia (increased curvature, decreased diameter), particularly in Weill-Marchesani syndrome.
 Associated pupil abnormality in ectopia lentis et pupillae (autosomal recessive inheritance), classically displacement in opposite direction (typically downward) from the lens (typically upward), but showing much variation even between eyes and among family members. In MFS pupils often dilate poorly.
Associated pupil abnormality in ectopia lentis et pupillae (autosomal recessive inheritance), classically displacement in opposite direction (typically downward) from the lens (typically upward), but showing much variation even between eyes and among family members. In MFS pupils often dilate poorly.
 Glaucoma may result acutely from pupillary block by anteriorly displaced lens (especially with homocystinuria or Weill-Marchesani syndrome). Ectopia lentis may be a consequence of buphthalmos or megalocornea, due to mechanical stretching of zonule. Corneal enlargement with normal IOP is also occasionally an association of ectopia lentis, regardless of origin.
Glaucoma may result acutely from pupillary block by anteriorly displaced lens (especially with homocystinuria or Weill-Marchesani syndrome). Ectopia lentis may be a consequence of buphthalmos or megalocornea, due to mechanical stretching of zonule. Corneal enlargement with normal IOP is also occasionally an association of ectopia lentis, regardless of origin.
 Cataract develops prematurely in most dislocated lenses, and there is significant increased risk of rhegmatogenous retinal detachment, possibly greater after lens extraction surgery. These complications are seldom seen in childhood.
Cataract develops prematurely in most dislocated lenses, and there is significant increased risk of rhegmatogenous retinal detachment, possibly greater after lens extraction surgery. These complications are seldom seen in childhood.
 Refractive error (in the phakic portion of pupil) combines lenticular myopia (from steepened overall lens curvature) and astigmatism (optical power near lens edge least for axis perpendicular to edge, greatest for axis parallel to edge tangent). Many eyes with ectopia lentis (in MFS as well as other disorders) also have a significant component of axial myopia. Myopia may be the first sign of ectopia lentis, evident before displacement of the lens becomes obvious.
Refractive error (in the phakic portion of pupil) combines lenticular myopia (from steepened overall lens curvature) and astigmatism (optical power near lens edge least for axis perpendicular to edge, greatest for axis parallel to edge tangent). Many eyes with ectopia lentis (in MFS as well as other disorders) also have a significant component of axial myopia. Myopia may be the first sign of ectopia lentis, evident before displacement of the lens becomes obvious.
 With lens edge near the pupil center, retinal superimposition of phakic and aphakic images (one of which must be severely defocused) may degrade acuity to as low as 20/100 with best correction of either type, but in symmetrical cases significant amblyopia seldom occurs.
With lens edge near the pupil center, retinal superimposition of phakic and aphakic images (one of which must be severely defocused) may degrade acuity to as low as 20/100 with best correction of either type, but in symmetrical cases significant amblyopia seldom occurs.
FIGURE 8.10. Dislocated lenses. A: Traumatic dislocation with cataract in a physically abused infant; rhegmatogenous retinal detachment from a giant tear was also present in both eyes. B: Upward displacement in an older child with Marfan syndrome.
Systemic Findings
 MFS (autosomal dominant inheritance, mutation of fibrillin gene—FBN1—chromosome 15q21) is defined by characteristic cardiovascular abnormalities (aortic root dilation with valvular insufficiency, mitral valve prolapse, risk of dissecting aneurysm) and body habitus (tall stature, arachnodactyly, hyperextensible joints).
MFS (autosomal dominant inheritance, mutation of fibrillin gene—FBN1—chromosome 15q21) is defined by characteristic cardiovascular abnormalities (aortic root dilation with valvular insufficiency, mitral valve prolapse, risk of dissecting aneurysm) and body habitus (tall stature, arachnodactyly, hyperextensible joints).
 Homocystinuria (autosomal recessive, deficiency of cystethione beta-synthase, with increased blood and urine methionine) is also associated with tall stature and other skeletal abnormalities, but tight joints. Mental deficiency and deterioration may be reduced by diet low in methionine, or in some milder cases by supplementation with pyridoxine (vitamin B6). A major concern is high risk of thromboembolism, particularly in association with general anesthesia.
Homocystinuria (autosomal recessive, deficiency of cystethione beta-synthase, with increased blood and urine methionine) is also associated with tall stature and other skeletal abnormalities, but tight joints. Mental deficiency and deterioration may be reduced by diet low in methionine, or in some milder cases by supplementation with pyridoxine (vitamin B6). A major concern is high risk of thromboembolism, particularly in association with general anesthesia.
 Weill-Marchesani syndrome (autosomal recessive inheritance) is characterized by short stature, brachycephaly, and brachydactyly.
Weill-Marchesani syndrome (autosomal recessive inheritance) is characterized by short stature, brachycephaly, and brachydactyly.
 Isolated ectopia lentis may be sporadic or show a familial pattern that is recessive or dominant (often caused by an FBN1 mutation different from those responsible for MFS).
Isolated ectopia lentis may be sporadic or show a familial pattern that is recessive or dominant (often caused by an FBN1 mutation different from those responsible for MFS).
Treatment and Management
 Refractive correction for myopia and/or astigmatism. If the lens edge is displaced to near the center of the pupil, aphakic refractive correction may be preferred.
Refractive correction for myopia and/or astigmatism. If the lens edge is displaced to near the center of the pupil, aphakic refractive correction may be preferred.
 Lens removal is seldom necessary for visual reasons and may be associated with serious complications. Use of suture-supported lens implants remains controversial.
Lens removal is seldom necessary for visual reasons and may be associated with serious complications. Use of suture-supported lens implants remains controversial.
Congenital and Developmental Cataract
Summary
Congenital (present at birth) and developmental (onset during infancy or childhood) cataract may affect one or both eyes. Unilateral congenital cataract usually reflects localized maldevelopment of ocular structures. Bilateral cataract in childhood is often familial (Figure 8.11A), or associated with a systemic disorder, and should prompt work-up for an underlying cause. (Table 8.11 provides a partial listing; see also discussion of differential diagnosis below.)
FIGURE 8.11. Developmental cataracts. A: Familial lamellar cataract, onset at age 3 years. Note central light transmission through clear nucleus. Amblyopia is rarely caused by this type of bilateral cataract. B: Posterior lenticonus with early PSC opacification.
Diagnostic Evaluation
 Critical assessment of lens and cataract morphology, with close attention to localization of opacity (Table 8.12). Of particular importance in unilateral cases is the finding of posterior lenticonus, thinning of the posterior capsule, usually central, that is associated with posterior bulging of cortex. Opacification, beginning at the posterior pole and often extending to the nucleus, may not develop until after infancy. Prior to cataract formation, myopia may result from steepened curvature of affected posterior capsule (Figure 8.11B).
Critical assessment of lens and cataract morphology, with close attention to localization of opacity (Table 8.12). Of particular importance in unilateral cases is the finding of posterior lenticonus, thinning of the posterior capsule, usually central, that is associated with posterior bulging of cortex. Opacification, beginning at the posterior pole and often extending to the nucleus, may not develop until after infancy. Prior to cataract formation, myopia may result from steepened curvature of affected posterior capsule (Figure 8.11B).
 Comprehensive eye examination to look for associated developmental or degenerative abnormalities, including microcornea or microphthalmos (Tables 8.5 and 8.6), anterior segment dysgenesis (Table 8.8), persistent hyperplastic primary vitreous (PHPV) and other vitreoretinal disorders (p. 452), and pigmentary retinopathy (p. 451, Table 8.15). B-scan ultrasonography if fundus visualization is inadequate.
Comprehensive eye examination to look for associated developmental or degenerative abnormalities, including microcornea or microphthalmos (Tables 8.5 and 8.6), anterior segment dysgenesis (Table 8.8), persistent hyperplastic primary vitreous (PHPV) and other vitreoretinal disorders (p. 452), and pigmentary retinopathy (p. 451, Table 8.15). B-scan ultrasonography if fundus visualization is inadequate.
 With or without a history of maternal illness during pregnancy, serologic testing for congenital infection, including toxoplasmosis, rubella, syphilis, cytomegalovirus, and HSV (TORCH titers) is justified in infants with unexplained lens opacification (unilateral or bilateral), but only rubella (rare in North America in recent decades—see below) typically causes visually significant cataract.
With or without a history of maternal illness during pregnancy, serologic testing for congenital infection, including toxoplasmosis, rubella, syphilis, cytomegalovirus, and HSV (TORCH titers) is justified in infants with unexplained lens opacification (unilateral or bilateral), but only rubella (rare in North America in recent decades—see below) typically causes visually significant cataract.
 Blood chemistry testing for glucose, calcium: Both hypoglycemia and hypocalcemia may cause lens opacification. Diabetic cataract occurs rarely in childhood.
Blood chemistry testing for glucose, calcium: Both hypoglycemia and hypocalcemia may cause lens opacification. Diabetic cataract occurs rarely in childhood.
 Urine testing for reducing substance (after milk feeding, to look for galactosemia), amino acids (Lowe syndrome—test may be negative during first few months of life). (See below.)
Urine testing for reducing substance (after milk feeding, to look for galactosemia), amino acids (Lowe syndrome—test may be negative during first few months of life). (See below.)
 Comprehensive evaluation by pediatrician or genetics specialist for associated dysmorphic or systemic abnormalities (e.g., distinctive small beaklike nose in Hallermann-Streiff syndrome; characteristic skin and skeletal findings in Conradi syndrome), which may prompt additional testing (chromosome analysis, neuroimaging, etc.) Usually none found in a healthy appearing infant.
Comprehensive evaluation by pediatrician or genetics specialist for associated dysmorphic or systemic abnormalities (e.g., distinctive small beaklike nose in Hallermann-Streiff syndrome; characteristic skin and skeletal findings in Conradi syndrome), which may prompt additional testing (chromosome analysis, neuroimaging, etc.) Usually none found in a healthy appearing infant.
 Examine eyes of parents and siblings: many childhood cataracts are familial. Female relatives of boys with Lowe syndrome may show typical carrier state findings.
Examine eyes of parents and siblings: many childhood cataracts are familial. Female relatives of boys with Lowe syndrome may show typical carrier state findings.
 Many mutations involving various genes have been identified as causes of isolated hereditary cataract, mostly with autosomal dominant transmission. Some cause highly distinctive and uniform congenital or developmental lens changes, while others show variable expression in terms of morphology, extent of opacification, and time course.
Many mutations involving various genes have been identified as causes of isolated hereditary cataract, mostly with autosomal dominant transmission. Some cause highly distinctive and uniform congenital or developmental lens changes, while others show variable expression in terms of morphology, extent of opacification, and time course.
Table 8.11 Important systemic associations of cataract in childhood
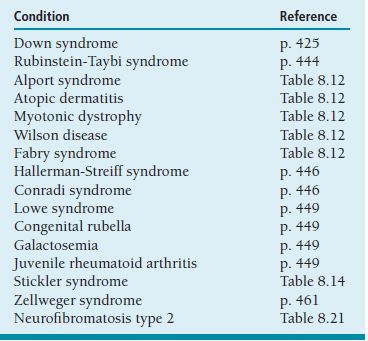
Table 8.12 Types of pediatric cataract
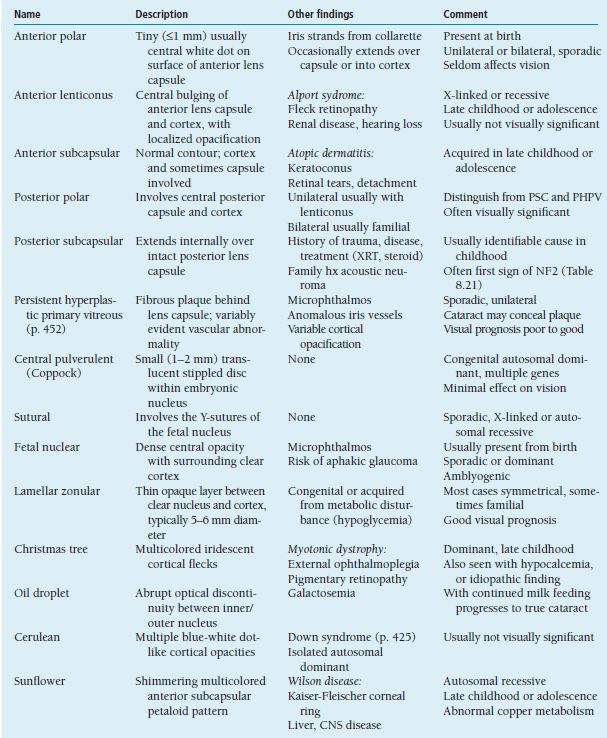
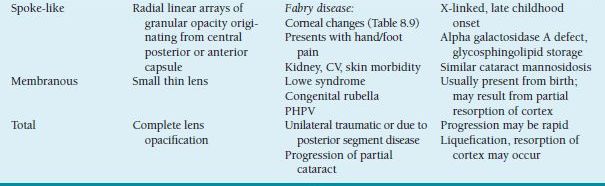
CNS, central nervous system; CV, cardiovascular; NF2, neurofibromatosis type 2; PHPV, persistent hyperplastic primary vitreous PSC, posterior subcapsular; XRT, x-ray therapy.
Treatment Considerations
 Dense (i.e., completely blocking light transmission) axial opacities more than 3 mm in diameter are likely to cause severe amblyopia within months of onset in infancy or early childhood, and require prompt surgical treatment whether bilateral or unilateral. Cataracts that permit light transmission centrally or that are smaller than 3 mm should be monitored closely, provided there is no development of nystagmus or nonalternating constant strabismus indicating possibly serious visual deprivation.
Dense (i.e., completely blocking light transmission) axial opacities more than 3 mm in diameter are likely to cause severe amblyopia within months of onset in infancy or early childhood, and require prompt surgical treatment whether bilateral or unilateral. Cataracts that permit light transmission centrally or that are smaller than 3 mm should be monitored closely, provided there is no development of nystagmus or nonalternating constant strabismus indicating possibly serious visual deprivation.
 Cataract extraction performed before age 6 years should include routine posterior capsulectomy and vitrectomy because of the high frequency of secondary opacification at young ages.
Cataract extraction performed before age 6 years should include routine posterior capsulectomy and vitrectomy because of the high frequency of secondary opacification at young ages.
 Primary intraocular lens implantation is now well accepted as appropriate and even desirable in conjunction with unilateral cataract surgery in most children over age 2 years, and possibly as young as 6 months. To help ensure optimal visual development during the critical early years of life, IOL power selection should bring the eye as close to emmetropia as possible in the early postoperative period, recognizing the likelihood of significant myopic shift over time (see below). Close follow-up with obsessive attention to refractive and occlusion therapy needs over many years is essential to achieving best possible vision.
Primary intraocular lens implantation is now well accepted as appropriate and even desirable in conjunction with unilateral cataract surgery in most children over age 2 years, and possibly as young as 6 months. To help ensure optimal visual development during the critical early years of life, IOL power selection should bring the eye as close to emmetropia as possible in the early postoperative period, recognizing the likelihood of significant myopic shift over time (see below). Close follow-up with obsessive attention to refractive and occlusion therapy needs over many years is essential to achieving best possible vision.
 Primary intraocular lens implantation is an option for bilateral cataract surgery in older children, but has not been shown to improve visual outcomes. Deciding whether a child is best managed with primary bilateral IOL implantation or with surgery that leaves the aphakic eyes well positioned for possible future secondary implantation requires careful consideration of options with the family.
Primary intraocular lens implantation is an option for bilateral cataract surgery in older children, but has not been shown to improve visual outcomes. Deciding whether a child is best managed with primary bilateral IOL implantation or with surgery that leaves the aphakic eyes well positioned for possible future secondary implantation requires careful consideration of options with the family.
 Currently available evidence does not support the use of primary intraocular lens implantation in infants with bilateral cataract, or less than 6 months old. Lensectomy with vitrectomy should be performed in such patients, leaving sufficient peripheral lens capsule to support possible future secondary IOL implantation. Aphakic refractive correction is provided in the form of extended wear silicone contact lenses or high plus spectacle lenses.
Currently available evidence does not support the use of primary intraocular lens implantation in infants with bilateral cataract, or less than 6 months old. Lensectomy with vitrectomy should be performed in such patients, leaving sufficient peripheral lens capsule to support possible future secondary IOL implantation. Aphakic refractive correction is provided in the form of extended wear silicone contact lenses or high plus spectacle lenses.
Disease Course
 While subnormal axial length (microphthalmos) is common with congenital cataract, when treatment is delayed excessive growth causing axial elongation and myopic refractive shift may result from visual deprivation. (Transient media opacities such as corneal clouding or vitreous hemorrhage from birth trauma may have similar effect.) After surgery in infancy, myopic shift (evident as decreased plus requirement in aphakic eyes) typically occurs as a result of normal growth of the eye, though the amount is highly variable and unpredictable.
While subnormal axial length (microphthalmos) is common with congenital cataract, when treatment is delayed excessive growth causing axial elongation and myopic refractive shift may result from visual deprivation. (Transient media opacities such as corneal clouding or vitreous hemorrhage from birth trauma may have similar effect.) After surgery in infancy, myopic shift (evident as decreased plus requirement in aphakic eyes) typically occurs as a result of normal growth of the eye, though the amount is highly variable and unpredictable.
 Open angle glaucoma is a common development following pediatric cataract surgery, requiring careful monitoring of IOP in aphakic and pseudophakic children. Risk is greatest in microphthalmic eyes with congenital nuclear or total cataract, or PHPV. Onset is usually insidious, often more than 5 years after surgery, with more rapid than expected myopic shift in refraction as the first sign. May be challenging to treat, frequently requiring multiple surgeries, though in some cases control with topical medication is possible.
Open angle glaucoma is a common development following pediatric cataract surgery, requiring careful monitoring of IOP in aphakic and pseudophakic children. Risk is greatest in microphthalmic eyes with congenital nuclear or total cataract, or PHPV. Onset is usually insidious, often more than 5 years after surgery, with more rapid than expected myopic shift in refraction as the first sign. May be challenging to treat, frequently requiring multiple surgeries, though in some cases control with topical medication is possible.
 Pupillary block with pressure elevation may occur following lensectomy/vitrectomy in early infancy, as a result of regrowth of vitreous and extension into the anterior chamber. Peripheral iridectomy should accompany L/V surgery performed before age 6 months.
Pupillary block with pressure elevation may occur following lensectomy/vitrectomy in early infancy, as a result of regrowth of vitreous and extension into the anterior chamber. Peripheral iridectomy should accompany L/V surgery performed before age 6 months.
 Retinal detachment, once a frequent and feared late (often decades after surgery) complication of pediatric cataract surgery, seems much less a concern with modern operative techniques. Clinically significant cystoid macular edema is rarely seen after uncomplicated cataract surgery in childhood.
Retinal detachment, once a frequent and feared late (often decades after surgery) complication of pediatric cataract surgery, seems much less a concern with modern operative techniques. Clinically significant cystoid macular edema is rarely seen after uncomplicated cataract surgery in childhood.
Differential Diagnosis
 Lowe syndrome (oculocerebrorenal syndrome) is an X-linked recessive disorder that nearly always presents with bilateral congenital cataract. The lens is typically small, thin, and totally opacified. Glaucoma develops during infancy in slightly more than half of cases. Renal tubular dysfunction causes metabolic acidosis and aminoaciduria; rarely renal failure. Mental deficiency ranges from mild to severe.
Lowe syndrome (oculocerebrorenal syndrome) is an X-linked recessive disorder that nearly always presents with bilateral congenital cataract. The lens is typically small, thin, and totally opacified. Glaucoma develops during infancy in slightly more than half of cases. Renal tubular dysfunction causes metabolic acidosis and aminoaciduria; rarely renal failure. Mental deficiency ranges from mild to severe.
 Congenital rubella (Gregg syndrome) is associated with cataract in up to one third of cases (75% bilateral), and glaucoma in about 10%. Pigmentary retinopathy (“salt and pepper” fundus) is seen in nearly half of cases, usually having only minor impact on vision. Important systemic findings include deafness, mental deficiency, hepatosplenomegaly, and thrombocytopenia. Live virus has been found in lens aspirates several years after birth, necessitating infection control precautions during surgery and contributing to frequently severe postoperative inflammation.
Congenital rubella (Gregg syndrome) is associated with cataract in up to one third of cases (75% bilateral), and glaucoma in about 10%. Pigmentary retinopathy (“salt and pepper” fundus) is seen in nearly half of cases, usually having only minor impact on vision. Important systemic findings include deafness, mental deficiency, hepatosplenomegaly, and thrombocytopenia. Live virus has been found in lens aspirates several years after birth, necessitating infection control precautions during surgery and contributing to frequently severe postoperative inflammation.
 High blood levels of galactose lead to infantile onset of cataract in patients with the autosomal recessive disorders galactosemia (deficiency of galactose-1-phosphate uridyl transferase—GALT) and galactokinase (GALK) deficiency, both diagnosed by assay of enzyme activity in RBCs. Prompt initiation of a soy formula diet may reverse early lens changes (beginning with the classic “oil droplet” appearance, described in Table 8.12), as well as systemic consequences (failure to thrive, signs of liver disease, mental deficiency, and often sepsis) associated with GALT galactosemia only. In North America, most cases are identified through state-mandated newborn screening programs, but both false-negative and false-positive screening results are common.
High blood levels of galactose lead to infantile onset of cataract in patients with the autosomal recessive disorders galactosemia (deficiency of galactose-1-phosphate uridyl transferase—GALT) and galactokinase (GALK) deficiency, both diagnosed by assay of enzyme activity in RBCs. Prompt initiation of a soy formula diet may reverse early lens changes (beginning with the classic “oil droplet” appearance, described in Table 8.12), as well as systemic consequences (failure to thrive, signs of liver disease, mental deficiency, and often sepsis) associated with GALT galactosemia only. In North America, most cases are identified through state-mandated newborn screening programs, but both false-negative and false-positive screening results are common.
References
Francis PJ, Berry V, Bhattacharya SS, et al. The genetics of childhood cataract. J Med Genet 2000;37:481–488.
Greenwald MJ, Glaser SR. Visual outcomes after surgery for unilateral cataract in children over two years old: posterior chamber intraocular lens implantation versus contact lens correction of aphakia. J AAPOS 1998;2:168–176.
Juvenile Rheumatoid Arthritis–Associated Uveitis
Summary
By far the most common and important form of pediatric uveitis is that seen with juvenile rheumatoid arthritis (JRA), typically presenting in girls under 16 years old.
Demographics
 Fifty percent or more of JRA cases are pauciarticular, and 20% of these develop uveitis.
Fifty percent or more of JRA cases are pauciarticular, and 20% of these develop uveitis.
 Female-to-male ratio is 3:1 or greater.
Female-to-male ratio is 3:1 or greater.
 Onset of arthritis before age 7 years is a risk factor for development of uveitis.
Onset of arthritis before age 7 years is a risk factor for development of uveitis.
Symptoms and Signs
 Blurred vision or floaters.
Blurred vision or floaters.
 Ocular redness and discomfort typically absent to mild; occasionally more severe.
Ocular redness and discomfort typically absent to mild; occasionally more severe.
 Most patients have no ocular symptoms and are diagnosed after failing a vision test or when screened after onset of joint symptoms leads to systemic diagnosis.
Most patients have no ocular symptoms and are diagnosed after failing a vision test or when screened after onset of joint symptoms leads to systemic diagnosis.
Ophthalmic Findings
 Bilateral (75% of cases) or unilateral anterior nongranulomatous uveitis.
Bilateral (75% of cases) or unilateral anterior nongranulomatous uveitis.
 Anterior chamber cell reaction and keratic precipitate formation are both generally of mild to moderate degree with few vitreous cells. (Perform careful slit-lamp examination using the brightest available light setting.)
Anterior chamber cell reaction and keratic precipitate formation are both generally of mild to moderate degree with few vitreous cells. (Perform careful slit-lamp examination using the brightest available light setting.)
 Posterior synechiae and band keratopathy are important early indicators of tissue damage.
Posterior synechiae and band keratopathy are important early indicators of tissue damage.
 Cataract, glaucoma, and macular edema are late sight-threatening complications.
Cataract, glaucoma, and macular edema are late sight-threatening complications.
Systemic Findings
 Arthritis usually involves a small number of larger joints (fewer than 5, especially the knees), but uveitis occasionally occurs in polyarticular or systemic JRA.
Arthritis usually involves a small number of larger joints (fewer than 5, especially the knees), but uveitis occasionally occurs in polyarticular or systemic JRA.
 Uveitis may precede or follow joint involvement, or occur in its absence.
Uveitis may precede or follow joint involvement, or occur in its absence.
Special Tests
 ANA strongly positive in a majority of cases.
ANA strongly positive in a majority of cases.
 Rheumatoid factor usually negative.
Rheumatoid factor usually negative.
 HLA-B27 may be positive. Uveitis is more likely to be acute and symptomatic in this group, which tends also to be older and includes more males.
HLA-B27 may be positive. Uveitis is more likely to be acute and symptomatic in this group, which tends also to be older and includes more males.
Treatment and Management
 Mydriatic drops and topical or periocular injected corticosteroid are used to minimize anterior chamber cell reaction and halt progression of posterior synechiae.
Mydriatic drops and topical or periocular injected corticosteroid are used to minimize anterior chamber cell reaction and halt progression of posterior synechiae.
 Vision loss from macular edema requires periocular injection of long-acting corticosteroid (e.g., triamcinolone) or systemic steroid treatment in most cases.
Vision loss from macular edema requires periocular injection of long-acting corticosteroid (e.g., triamcinolone) or systemic steroid treatment in most cases.
 Immunosuppressive treatment (e.g., methotrexate) may be necessary in refractory cases.
Immunosuppressive treatment (e.g., methotrexate) may be necessary in refractory cases.
 IOP may be elevated from the inflammatory process or effect of steroid treatment; must be monitored closely.
IOP may be elevated from the inflammatory process or effect of steroid treatment; must be monitored closely.
 Cataract may progress to require surgery, with significantly increased frequency of complications. Inflammation should be quiescent or maximally suppressed during the perioperative period. Intraocular lens implantation is controversial; may be appropriate in carefully selected cases.
Cataract may progress to require surgery, with significantly increased frequency of complications. Inflammation should be quiescent or maximally suppressed during the perioperative period. Intraocular lens implantation is controversial; may be appropriate in carefully selected cases.
Differential Diagnosis
 Both childhood sarcoidosis and juvenile psoriatic arthritis may present with chronic anterior uveitis and joint inflammation, sometimes before development of characteristic lesions of skin or other organs, and may be mistaken for JRA-associated uveitis. Bilateral hilar adenopathy is nearly always present in children with sarcoidosis.
Both childhood sarcoidosis and juvenile psoriatic arthritis may present with chronic anterior uveitis and joint inflammation, sometimes before development of characteristic lesions of skin or other organs, and may be mistaken for JRA-associated uveitis. Bilateral hilar adenopathy is nearly always present in children with sarcoidosis.
 Nongranulomatous anterior uveitis (sometimes very intense but usually responding well to treatment) may accompany tubulointerstitial nephritis (TINU syndrome) in children (mostly girls). Abnormal laboratory findings include proteinuria, elevated serum creatinine, and high ESR.
Nongranulomatous anterior uveitis (sometimes very intense but usually responding well to treatment) may accompany tubulointerstitial nephritis (TINU syndrome) in children (mostly girls). Abnormal laboratory findings include proteinuria, elevated serum creatinine, and high ESR.
 Kawasaki disease is an acute vasculitic disorder in infancy or childhood, one of whose principal manifestations is conjunctival hyperemia (without discharge), typically in a context of febrile illness and skin rash. Uveitis (nearly always mild) and joint pain/swelling may raise concern about JRA. Other findings include erythema and peeling/splitting of palms/soles/lips. Prevention of life-threatening coronary artery aneurysm, the most serious complication, requires aggressive pediatric medical management including IV immunoglobulin and high-dose aspirin.
Kawasaki disease is an acute vasculitic disorder in infancy or childhood, one of whose principal manifestations is conjunctival hyperemia (without discharge), typically in a context of febrile illness and skin rash. Uveitis (nearly always mild) and joint pain/swelling may raise concern about JRA. Other findings include erythema and peeling/splitting of palms/soles/lips. Prevention of life-threatening coronary artery aneurysm, the most serious complication, requires aggressive pediatric medical management including IV immunoglobulin and high-dose aspirin.
References
Kanski JJ, Petty RE. Chronic childhood arthritis and uveitis. Ocular Infect Immunol 1996;40:485–493.
Lam LA, Lowder CY, Baervelt G, et al. Surgical management of cataract in children with juvenile rheumatoid arthritis. Am J Ophthalmol 2003;125:772–778.
Intermediate Uveitis (Pars Planitis)
Summary
The term intermediate uveitis is preferred for designation of chronic intraocular inflammation concentrated in the anterior vitreous.
Demographics
 Twenty-five percent of pediatric uveitis cases, second in frequency after JRA.
Twenty-five percent of pediatric uveitis cases, second in frequency after JRA.
 Bilateral in 75% of cases.
Bilateral in 75% of cases.
Symptoms and Signs
 Decreased vision, floaters.
Decreased vision, floaters.
 Redness and discomfort rarely present; often diagnosed after failure on routine vision testing.
Redness and discomfort rarely present; often diagnosed after failure on routine vision testing.
Ophthalmic Findings
 Mild anterior chamber cells and flare, usually no keratic precipitates
Mild anterior chamber cells and flare, usually no keratic precipitates
 Posterior synechiae and band keratopathy much less common than with JRA.
Posterior synechiae and band keratopathy much less common than with JRA.
 Anterior vitreous cells always present, usually far in excess of aqueous cells.
Anterior vitreous cells always present, usually far in excess of aqueous cells.
 Posterior subcapsular (PSC) cataract common; often remains mild but may progress to require surgical treatment.
Posterior subcapsular (PSC) cataract common; often remains mild but may progress to require surgical treatment.
 Peripheral fundus examination (with scleral indentation if possible; employing sedation or general anesthesia if necessary) is essential to identify characteristic pars plana “snow banking,” retinal vasculitis in a minority of cases, and occasionally a localized chorioretinal lesion that may indicate an infectious process (e.g., toxoplasmosis).
Peripheral fundus examination (with scleral indentation if possible; employing sedation or general anesthesia if necessary) is essential to identify characteristic pars plana “snow banking,” retinal vasculitis in a minority of cases, and occasionally a localized chorioretinal lesion that may indicate an infectious process (e.g., toxoplasmosis).
 Macular edema is the most frequent cause of vision loss.
Macular edema is the most frequent cause of vision loss.
 Less common complications include retinal neovascularization, retinal detachment, and glaucoma.
Less common complications include retinal neovascularization, retinal detachment, and glaucoma.
Special Tests
 Fluorescein angiography to look for macular edema and vasculitis.
Fluorescein angiography to look for macular edema and vasculitis.
 Chest x-ray to look for evidence of tuberculosis and sarcoidosis.
Chest x-ray to look for evidence of tuberculosis and sarcoidosis.
 Lyme disease titers if the patient has a history of possible tick bite or exposure. Intermediate uveitis is the most common intraocular manifestation of this condition. Other findings may include cranial nerve palsies (especially facial), optic neuritis, keratitis, and conjunctivitis (associated with characteristic skin rash in the acute phase).
Lyme disease titers if the patient has a history of possible tick bite or exposure. Intermediate uveitis is the most common intraocular manifestation of this condition. Other findings may include cranial nerve palsies (especially facial), optic neuritis, keratitis, and conjunctivitis (associated with characteristic skin rash in the acute phase).
Treatment and Management
 Topical and periocular corticosteroids; mydriatic drops if forming synechiae.
Topical and periocular corticosteroids; mydriatic drops if forming synechiae.
 Monitor closely for vision loss from macular edema.
Monitor closely for vision loss from macular edema.
 Systemic immunosuppression may be necessary in refractory cases.
Systemic immunosuppression may be necessary in refractory cases.
 Pars plana cryotherapy and vitrectomy are sometimes performed in patients with severe disease.
Pars plana cryotherapy and vitrectomy are sometimes performed in patients with severe disease.
Toxocariasis
Summary
Ocular toxocariasis occurs almost exclusively in children and may present either with intense posterior segment inflammation or as a cicatricial lesion involving retina and vitreous.
Demographics
 Average age of presentation is 7 years.
Average age of presentation is 7 years.
 Mostly boys.
Mostly boys.
Etiology
Children ingest eggs of the roundworm Toxocara canis with soil contaminated by dog feces; larvae develop in the intestines and disseminate via hematogenous spread throughout the body, including the posterior segment of the eye. Inflammation may increase after death of the organism.
Symptoms and Signs
 Decreased vision, floaters; nearly always unilateral.
Decreased vision, floaters; nearly always unilateral.
 Mild redness and discomfort in some acute cases.
Mild redness and discomfort in some acute cases.
 May present with leukocoria that resembles retinoblastoma (p. 481).
May present with leukocoria that resembles retinoblastoma (p. 481).
Ophthalmic Findings
 In chronic cases, the usual lesion is peripheral fundus or posterior pole granuloma, an elevated whitish mass, often associated with localized tractional retinal detachment, and sometimes mild overlying vitreous cell reaction. May resemble PHPV (p. 452).
In chronic cases, the usual lesion is peripheral fundus or posterior pole granuloma, an elevated whitish mass, often associated with localized tractional retinal detachment, and sometimes mild overlying vitreous cell reaction. May resemble PHPV (p. 452).
 Vitreitis in acute cases may be so severe it prevents visualization of the fundus. Anterior chamber reaction usually mild if present but occasionally there is hypopyon.
Vitreitis in acute cases may be so severe it prevents visualization of the fundus. Anterior chamber reaction usually mild if present but occasionally there is hypopyon.
 Rarely presents with neuroretinitis or optic neuropathy.
Rarely presents with neuroretinitis or optic neuropathy.
 Macular edema, cataract, and glaucoma are occasional complications.
Macular edema, cataract, and glaucoma are occasional complications.
Systemic Findings
 Usually absent in children with ocular disease.
Usually absent in children with ocular disease.
 The syndrome of visceral larvae migrans, with fever, respiratory and neurologic symptoms, and peripheral blood eosinophilia, is typically seen in children under age 3 years, without ocular involvement.
The syndrome of visceral larvae migrans, with fever, respiratory and neurologic symptoms, and peripheral blood eosinophilia, is typically seen in children under age 3 years, without ocular involvement.
Special Tests
 Enzyme-linked immunosorbent assay (ELISA) for T. canis is usually diagnostic.
Enzyme-linked immunosorbent assay (ELISA) for T. canis is usually diagnostic.
 CBC may show elevated eosinophil count.
CBC may show elevated eosinophil count.
Treatment and Management
 In consultation with an infectious disease specialist, consider systemic antihelminthic treatment.
In consultation with an infectious disease specialist, consider systemic antihelminthic treatment.
 Periocular or systemic corticosteroid treatment for ocular inflammation, which may increase after antihelminthic therapy.
Periocular or systemic corticosteroid treatment for ocular inflammation, which may increase after antihelminthic therapy.
 Vitrectomy may be indicated for retinal detachment or vitreoretinal traction, or to remove immunogenic debris from the vitreous.
Vitrectomy may be indicated for retinal detachment or vitreoretinal traction, or to remove immunogenic debris from the vitreous.
Differential Diagnosis
 Intrauterine infection with the protozoan Toxoplasma gondii is the most common cause of posterior uveitis in infants and children, but intraocular inflammatory activity in the great majority of cases has subsided completely by the time of postnatal diagnosis of toxoplasmosis chorioretinitis.
Intrauterine infection with the protozoan Toxoplasma gondii is the most common cause of posterior uveitis in infants and children, but intraocular inflammatory activity in the great majority of cases has subsided completely by the time of postnatal diagnosis of toxoplasmosis chorioretinitis.
Disease results from maternal ingestion of raw meat or cat feces during pregnancy.
The usual ocular presentation is with one or more flat, sharply demarcated chorioretinal scars (characteristically showing both hyperpigmented and hypopigmented areas), typically involving the macula, often bilateral. A small minority of cases show more severe and extensive ocular damage, rarely leading to phthisis.
Reactivation of dormant organisms in retina, usually adjacent to a scar, may cause further loss of vision, so affected eyes need to be monitored carefully, and treated if active inflammation is found.
Central nervous system (CNS) involvement may cause seizures, hydrocephalus, and CT findings including loss of tissue and calcification, as well as hearing loss and mental deficiency.
Toxoplasmosis diagnosed in infancy or early childhood should generally receive aggressive systemic therapy with multiple drugs, primarily to minimize CNS damage, which may be progressive.
 Lymphocytic choriomeningitis virus infection is acquired from contact with rodent feces and may have severe effects on the fetus during pregnancy, including CNS damage and uveitis with chorioretinal scarring that may be indistinguishable from that of toxoplasmosis.
Lymphocytic choriomeningitis virus infection is acquired from contact with rodent feces and may have severe effects on the fetus during pregnancy, including CNS damage and uveitis with chorioretinal scarring that may be indistinguishable from that of toxoplasmosis.
 Other congenital infection (“ORCH” agents) involving the posterior segment is diagnosed much less often than toxoplasmosis. These conditions present with similar findings, though somewhat different typical patterns of fundus pigment disturbance (e.g., “salt and pepper” appearance in congenital rubella—p. 449) and brain calcification (e.g., periventricular deposits with congenital CMV infection). Active inflammatory chorioretinal lesions are rarely seen. Except in the case of HSV with cutaneous or ocular surface disease (p. 432), treatment is seldom indicated.
Other congenital infection (“ORCH” agents) involving the posterior segment is diagnosed much less often than toxoplasmosis. These conditions present with similar findings, though somewhat different typical patterns of fundus pigment disturbance (e.g., “salt and pepper” appearance in congenital rubella—p. 449) and brain calcification (e.g., periventricular deposits with congenital CMV infection). Active inflammatory chorioretinal lesions are rarely seen. Except in the case of HSV with cutaneous or ocular surface disease (p. 432), treatment is seldom indicated.
 Subacute sclerosing panencephalitis, a pediatric neurodegenerative disease caused by chronic measles virus infection, may present with evidence of chorioretinitis in the macular region.
Subacute sclerosing panencephalitis, a pediatric neurodegenerative disease caused by chronic measles virus infection, may present with evidence of chorioretinitis in the macular region.
 Posterior uveitis from noninfectious causes is very seldom seen in children.
Posterior uveitis from noninfectious causes is very seldom seen in children.
References
Gillespie SH, Dinning WJ, Voller A, et al. The spectrum of ocular toxocariasis. Eye 1993;7:415–418.
McLeod R, Boyer K, Karrison T, et al. Outcome of treatment for congenital toxoplasmosis, 1981–2004: the National Collaborative Chicago-Based Congenital Toxoplasmosis Study. Clin Infect Dis 2006;42:1383–1394.
Persistent Hyperplastic Primary Vitreous
Summary
PHPV, also known as persistent fetal vasculature, has a broad spectrum of severity with anterior and/or posterior segment involvement. There is usually a fibrous plaque on the posterior lens capsule associated with remnants of hyaloid (embryonic vitreous) structure extending from the optic nerve. Abnormal vascular elements are frequently but not always a part of the condition, which is sporadic, unilateral in nearly all cases, and without significant systemic associations.
Symptoms and Signs
 Decreased vision, often with secondary strabismus.
Decreased vision, often with secondary strabismus.
 Leukocoria variably evident (Figure 8.12A).
Leukocoria variably evident (Figure 8.12A).
 Smaller than normal eye.
Smaller than normal eye.
Ophthalmic Findings
 Microphthalmos to some degree always present, unless cornea has enlarged secondary to increased IOP in infancy.
Microphthalmos to some degree always present, unless cornea has enlarged secondary to increased IOP in infancy.
 Sometimes characteristic iris vascular abnormality, with vessels extending to pupil margin.
Sometimes characteristic iris vascular abnormality, with vessels extending to pupil margin.
 Variable lens opacification from none to total; often progressive over time. Opacified cortex may resorb leaving a membranous cataract.
Variable lens opacification from none to total; often progressive over time. Opacified cortex may resorb leaving a membranous cataract.
 Ciliary processes often dragged centrally into fibrovascular membrane adherent to posterior lens capsule. Anterior chamber may be shallow as a result of forward displacement of the lens and iris.
Ciliary processes often dragged centrally into fibrovascular membrane adherent to posterior lens capsule. Anterior chamber may be shallow as a result of forward displacement of the lens and iris.
 IOP may be normal, increased (due to anterior displacement of lens and iris), or decreased (due to traction on ciliary body).
IOP may be normal, increased (due to anterior displacement of lens and iris), or decreased (due to traction on ciliary body).
 Fibrovascular stalk extending from the optic disc to the posterior lens (usually central, sometimes peripheral) is common and very characteristic. May range from a slender thread to involvement of most of the vitreous.
Fibrovascular stalk extending from the optic disc to the posterior lens (usually central, sometimes peripheral) is common and very characteristic. May range from a slender thread to involvement of most of the vitreous.
 Significant posterior involvement, present in a minority of cases, typically takes the form of glial tissue covering a portion of the disc and adjacent retina, often with tractional distortion or elevation (Figure 8.12B), sometimes including a prominent fold.
Significant posterior involvement, present in a minority of cases, typically takes the form of glial tissue covering a portion of the disc and adjacent retina, often with tractional distortion or elevation (Figure 8.12B), sometimes including a prominent fold.
Special Tests
Ultrasound, CT, or MRI may reveal persistent posterior segment hyaloid structures obscured by anterior membrane or cataract.
Disease Course
Milder degrees often remain stable indefinitely. With greater severity, secondary developments including progressive lens opacification, displacement of lens and ciliary body, or vitreoretinal traction frequently lead to deterioration of vision, glaucoma or hypotony, retinal detachment, and ultimately phthisis. Posterior segment bleeding is an uncommon occurrence.
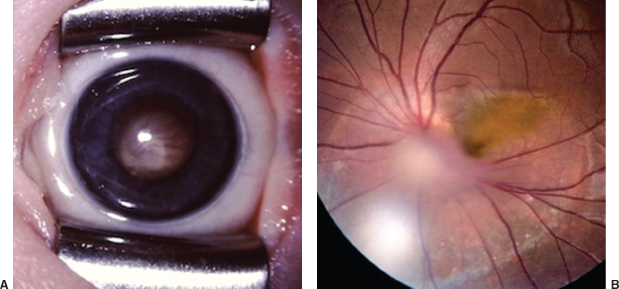
FIGURE 8.12. Persistent hyperplastic primary vitreous. A: Most of the posterior segment in this small eye is filled with abnormal fibrovascular tissue, causing shallowing of the anterior chamber, but lens remains clear. B: Posterior involvement in this eye (with 20/100 visual acuity) includes localized tractional elevation and distortion of peripapillary retina. The disc is partially obscured by fibrovascular tissue (out of focus in the image) extending toward the back of the lens, which remains clear.
Treatment and Management
 Mild cases may require no treatment, or simply part-time occlusion of the normal eye to reverse or prevent amblyopia.
Mild cases may require no treatment, or simply part-time occlusion of the normal eye to reverse or prevent amblyopia.
 Dense central retrolental plaque, alone or with secondary cataract, may be source of significant visual deprivation leading to severe amblyopia in infancy. Vitrectomy with lensectomy to restore media clarity, followed by contact lens correction of aphakia and occlusion for amblyopia may result in salvage of good or at least useful vision. IOL implantation is rarely an option because of age and microphthalmos.
Dense central retrolental plaque, alone or with secondary cataract, may be source of significant visual deprivation leading to severe amblyopia in infancy. Vitrectomy with lensectomy to restore media clarity, followed by contact lens correction of aphakia and occlusion for amblyopia may result in salvage of good or at least useful vision. IOL implantation is rarely an option because of age and microphthalmos.
 Surgery should be done via limbal incision; risk of retinal detachment is high after pars plana approach. Complete removal of the stalk is not necessary. Bleeding from abnormal vessels seldom a serious problem.
Surgery should be done via limbal incision; risk of retinal detachment is high after pars plana approach. Complete removal of the stalk is not necessary. Bleeding from abnormal vessels seldom a serious problem.
 When posterior segment involvement is severe, visual prognosis is poor but lensectomy may still be indicated to help stabilize IOP.
When posterior segment involvement is severe, visual prognosis is poor but lensectomy may still be indicated to help stabilize IOP.
Reference
Goldberg MF. Persistent fetal vasculature (PFV): an integrated interpretation of signs and symptoms associated with persistent hyperplastic primary vitreous (PHPV). Am J Ophthalmol 1997;124:587–626.
Vitreoretinal Dysplasia
Summary
Vitreoretinal dysplasia (VRD) describes severe maldevelopment of the posterior segment of the globe that results in replacement of most or all of the retina and vitreous by a mass of disorganized tissue, showing limited preservation of retinal differentiation. This condition is often part of a systemic syndrome (Table 8.13).
Symptoms and Signs
 Usually bilateral; unilateral cases may be difficult to distinguish from PHPV.
Usually bilateral; unilateral cases may be difficult to distinguish from PHPV.
 Severe vision loss in affected eyes.
Severe vision loss in affected eyes.
 Microphthalmos.
Microphthalmos.
 Leukocoria.
Leukocoria.
Special Tests
 Chromosome analysis.
Chromosome analysis.
 Genetic probes have been developed for several forms of retinal dysplasia, particularly Norrie disease in males.
Genetic probes have been developed for several forms of retinal dysplasia, particularly Norrie disease in males.
Differential Diagnosis
 End-stage retinopathy of prematurity (ROP) may closely resemble VRD, and in many cases of incontinentia pigmenti and Norrie disease the typical picture of VRD evolves during infancy through a vasculopathic process very similar to ROP (p. 457).
End-stage retinopathy of prematurity (ROP) may closely resemble VRD, and in many cases of incontinentia pigmenti and Norrie disease the typical picture of VRD evolves during infancy through a vasculopathic process very similar to ROP (p. 457).
 Fundus coloboma (p. 469) may be seen as a milder expression of retinal maldevelopment in trisomy 13 and Walker-Warburg syndrome.
Fundus coloboma (p. 469) may be seen as a milder expression of retinal maldevelopment in trisomy 13 and Walker-Warburg syndrome.
Table 8.13 Causes of vitreoretinal dysplasia
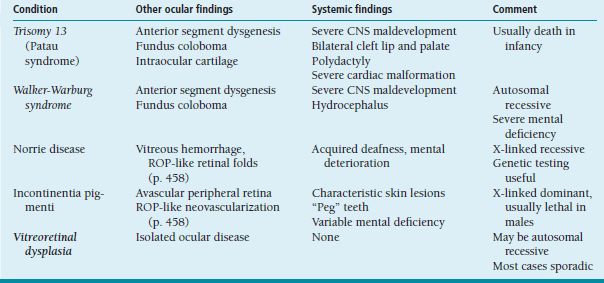
CNS, central nervous system; ROP, retinopathy of prematurity.
Vitreoretinal Degeneration
Liquefied or optically empty vitreous, sometimes with veils or hemorrhage, is associated with a variety of degenerative changes in the retina, and frequently early-onset cataract, in a number of hereditary disorders that typically manifest in childhood (Table 8.14). These conditions, particularly Stickler syndrome, are responsible for a large percentage of nontraumatic rhegmatogenous retinal detachments in the pediatric age range.
Retinopathy of Prematurity
Summary
ROP, formerly known as retrolental fibroplasia, is a disorder of retinal blood vessel development that occurs in infants born prematurely, prior to completion of retinal vascularization. It is characterized by neovascular proliferation in the transition zone between posterior vascularized retina and anterior avascular retina, which may cause permanent retinal damage by way of exudation or traction, and vision loss ranging from mild impairment to complete blindness.
Etiology
ROP is a complex multifactorial disorder, but considerable evidence supports the view that it originates with peripheral retinal hypoxia resulting from retinal tissue maturation in the face of arrested vascular development (at least to some degree a consequence of systemic hyperoxemia). This leads to release of vascular endothelial growth factor (VEGF), which stimulates proliferation of abnormal retinal vessels.
Risk Factors
 The incidence and severity of ROP are strongly inversely related to birth weight and gestational age. In the developed world, vision loss from ROP virtually never occurs following birth after 32 weeks’ gestation and at a weight greater than 2,000 g. A substantial majority of infants born before 28 weeks and weighing less than 1,000 g at birth will show at least mild transient ROP, and many will develop sight-threatening disease regardless of other circumstances or management.
The incidence and severity of ROP are strongly inversely related to birth weight and gestational age. In the developed world, vision loss from ROP virtually never occurs following birth after 32 weeks’ gestation and at a weight greater than 2,000 g. A substantial majority of infants born before 28 weeks and weighing less than 1,000 g at birth will show at least mild transient ROP, and many will develop sight-threatening disease regardless of other circumstances or management.
 Administration of supplemental oxygen in excess of clinical need has been well documented to increase the frequency and severity of ROP in vulnerable eyes. Even with carefully controlled oxygen administration, however, ROP is common in very low birth weight infants, and risk of severe vision loss cannot be eliminated by any strategy of oxygen curtailment that is compatible with optimal survival and neurologic health. There is no convincing evidence that excessive oxygen administration is harmful to the retina after 32 weeks’ gestational age, even when significant ROP is present.
Administration of supplemental oxygen in excess of clinical need has been well documented to increase the frequency and severity of ROP in vulnerable eyes. Even with carefully controlled oxygen administration, however, ROP is common in very low birth weight infants, and risk of severe vision loss cannot be eliminated by any strategy of oxygen curtailment that is compatible with optimal survival and neurologic health. There is no convincing evidence that excessive oxygen administration is harmful to the retina after 32 weeks’ gestational age, even when significant ROP is present.
 Risk factors of lesser importance include bronchopulmonary dysplasia, intraventricular hemorrhage, necrotizing enterocolitis, and sepsis.
Risk factors of lesser importance include bronchopulmonary dysplasia, intraventricular hemorrhage, necrotizing enterocolitis, and sepsis.
Table 8.14 Causes of vitreoretinal degeneration
ERG, electroretinogram; RD, retinal detachment.
Symptoms and Signs
 During the acute phase of ROP (roughly 1 to 6 months of age), when sight-saving clinical intervention is possible, ROP has no externally visible signs.
During the acute phase of ROP (roughly 1 to 6 months of age), when sight-saving clinical intervention is possible, ROP has no externally visible signs.
 Older infants and children with eyes that have been severely and irreversibly damaged by ROP may show leukocoria and microphthalmos. Strabismus and nystagmus may result from poor vision. End-stage ROP is characterized in some cases by inflammation and pain resulting from glaucoma in blind eyes.
Older infants and children with eyes that have been severely and irreversibly damaged by ROP may show leukocoria and microphthalmos. Strabismus and nystagmus may result from poor vision. End-stage ROP is characterized in some cases by inflammation and pain resulting from glaucoma in blind eyes.


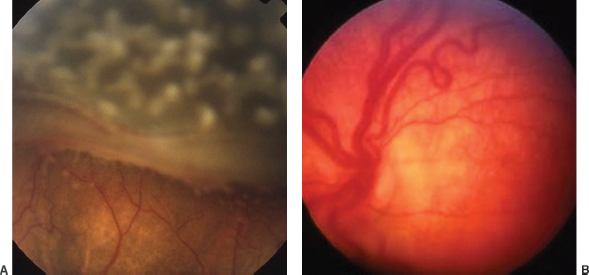
FIGURE 8.13. Acute-phase ROP. A: Stage 3 vasoproliferation, with extraretinal fibrovascular membrane arising from the posterior edge of a terminal ridge; peripheral avascular retina shows effect of recent laser treatment. B: Plus disease, with marked dilation of posterior venules and tortuosity of arterioles.
FIGURE 8.14. ROP with retinal detachment. A: Stage 4B with acute tractional and exudative detachment involving the macula. B: Same eye 13 years later, with persistent tractional detachment, and extensive pigment mottling after reabsorption of subretinal fluid. C: Florid fibrovascular proliferation extending to the back of the lens, leading to stage 5 funnel-shaped total detachment. D: Postmortem specimen showing funnel detachment.
Screening and Monitoring
 All infants born at a weight of less than 1,500 g, or at a gestational age of 30 weeks or less, must be screened for ROP. Screening is optional for infants 1,500 to 2,000 g and 31 weeks or more at birth who are judged by a responsible pediatric clinician to be at greater than usual risk.
All infants born at a weight of less than 1,500 g, or at a gestational age of 30 weeks or less, must be screened for ROP. Screening is optional for infants 1,500 to 2,000 g and 31 weeks or more at birth who are judged by a responsible pediatric clinician to be at greater than usual risk.
 Screening is done following dilation of the pupils with a mild mydriatic solution (usually 0.2% cyclopentolate combined with 1% phenylephrine [Cyclomydril]), by an experienced examiner using indirect ophthalmoscopy with the aid of a lid speculum and scleral indentation. A nurse or other skilled assistant should participate to monitor the infant’s vital signs, mitigate stress, and assist with positioning.
Screening is done following dilation of the pupils with a mild mydriatic solution (usually 0.2% cyclopentolate combined with 1% phenylephrine [Cyclomydril]), by an experienced examiner using indirect ophthalmoscopy with the aid of a lid speculum and scleral indentation. A nurse or other skilled assistant should participate to monitor the infant’s vital signs, mitigate stress, and assist with positioning.
 Initial screening is done at age 4 weeks or when gestational age has reached 31 weeks, whichever comes later. In the absence of positive findings, screening should be repeated at 1- to 3-week intervals (depending on risk level) at least until zone 2 is fully vascularized (i.e., vessels have reached the nasal ora serrata) and the infant has passed the gestational age of 35 weeks.
Initial screening is done at age 4 weeks or when gestational age has reached 31 weeks, whichever comes later. In the absence of positive findings, screening should be repeated at 1- to 3-week intervals (depending on risk level) at least until zone 2 is fully vascularized (i.e., vessels have reached the nasal ora serrata) and the infant has passed the gestational age of 35 weeks.
 Once ROP is present, its course should be monitored at intervals ranging from a few days to a few weeks (depending on severity and risk level) until complete disappearance of fundus abnormalities has occurred, or the infant has reached a developmental age of 45 to 50 weeks and shown no evidence of progression for at least 1 month.
Once ROP is present, its course should be monitored at intervals ranging from a few days to a few weeks (depending on severity and risk level) until complete disappearance of fundus abnormalities has occurred, or the infant has reached a developmental age of 45 to 50 weeks and shown no evidence of progression for at least 1 month.
Disease Course
 ROP is never present at birth. Onset usually occurs between ages 4 and 8 weeks, typically later for infants born at earlier gestational ages.
ROP is never present at birth. Onset usually occurs between ages 4 and 8 weeks, typically later for infants born at earlier gestational ages.
 Many eyes do not progress beyond stages 1 to 2. In most of these, the demarcation line or ridge gradually fades while migrating anteriorly toward the ora serrata.
Many eyes do not progress beyond stages 1 to 2. In most of these, the demarcation line or ridge gradually fades while migrating anteriorly toward the ora serrata.
 The initial development of extraretinal proliferation (stage 3 disease) or posterior vessel changes (preplus disease), both indicators of increased risk for vision loss, typically occurs 2 to 4 weeks after initial detection of a line or ridge but may not happen until as late as 42 weeks’ gestational age. From this point, the criterion for treatment is often reached within 1 to 2 weeks.
The initial development of extraretinal proliferation (stage 3 disease) or posterior vessel changes (preplus disease), both indicators of increased risk for vision loss, typically occurs 2 to 4 weeks after initial detection of a line or ridge but may not happen until as late as 42 weeks’ gestational age. From this point, the criterion for treatment is often reached within 1 to 2 weeks.
 ROP that begins in zone 1 or posterior zone 2 may progress rapidly and reach severity indicating a need for treatment within 1 to 2 weeks of initial detection. This is known as aggressive posterior ROP, formerly called rush disease.
ROP that begins in zone 1 or posterior zone 2 may progress rapidly and reach severity indicating a need for treatment within 1 to 2 weeks of initial detection. This is known as aggressive posterior ROP, formerly called rush disease.
 By the end of infancy, the lesions of acute phase ROP always regress or evolve into a state of scarring or cicatrization. Findings in cicatricial ROP may include the following:
By the end of infancy, the lesions of acute phase ROP always regress or evolve into a state of scarring or cicatrization. Findings in cicatricial ROP may include the following:
Persistent disturbances of retinal vasculature (usually minor, intraretinal) and pigmentation. Atrophic ERP membranes may separate from retinal surface and float in the vitreous.
Displacement of the macula (“dragging”), most often in the temporal direction, with decreased macular differentiation and loss of function to varying degree.
Persistent tractional retinal detachment (TRD) ranging from a narrow fold, often involving the macula, to total detachment showing a “closed funnel” configuration and a retrolental fibrovascular membrane.
Mild to moderate microphthalmos.
Myopia, often of high degree, with significant lenticular as well as axial components.
Glaucoma, resulting from anterior displacement of the iris and lens by retrolental cicatrix, in a minority of cases.
Treatment and Management
 Most eyes with ROP regress spontaneously, and no treatment is completely effective in preventing vision loss, or free of potentially serious complications. It is therefore standard practice to treat eyes only after ROP has reached a carefully defined level of severity.
Most eyes with ROP regress spontaneously, and no treatment is completely effective in preventing vision loss, or free of potentially serious complications. It is therefore standard practice to treat eyes only after ROP has reached a carefully defined level of severity.
 The multicenter CRYO-ROP study (1988) first confirmed that treatment of ROP significantly improves anatomic and functional outcomes. In this controlled randomized multicenter trial, cryotherapy was used to ablate the entire peripheral avascular retina when stage 3+ disease occupied at least 5 contiguous or 8 total clock hours of the fundus (“treatment threshold”).
The multicenter CRYO-ROP study (1988) first confirmed that treatment of ROP significantly improves anatomic and functional outcomes. In this controlled randomized multicenter trial, cryotherapy was used to ablate the entire peripheral avascular retina when stage 3+ disease occupied at least 5 contiguous or 8 total clock hours of the fundus (“treatment threshold”).
 Subsequent studies substituting laser ablation for cryotherapy have yielded similar or better results, making laser standard treatment in cases with sufficiently clear media. Inadequate response after initial treatment may improve following retreatment with laser (or cryo) to avascular retina that shows less than complete ablation. Close observation (1- to 2-week intervals or less) must be continued for at least 2 months after treatment.
Subsequent studies substituting laser ablation for cryotherapy have yielded similar or better results, making laser standard treatment in cases with sufficiently clear media. Inadequate response after initial treatment may improve following retreatment with laser (or cryo) to avascular retina that shows less than complete ablation. Close observation (1- to 2-week intervals or less) must be continued for at least 2 months after treatment.
 The ET-ROP multicenter study of earlier treatment for ROP (2003) showed greater benefit from laser treatment applied as soon as the presence of plus disease was documented, or when stage 3 was found present in zone 1. This is now the consensus criterion for treatment of ROP.
The ET-ROP multicenter study of earlier treatment for ROP (2003) showed greater benefit from laser treatment applied as soon as the presence of plus disease was documented, or when stage 3 was found present in zone 1. This is now the consensus criterion for treatment of ROP.
 The BEAT-ROP multicenter study (2011) recently confirmed that primary treatment with a single intravitreal injection of the VEGF inhibitor bevacizumab (Avastin) may be superior to laser for stage 3 ROP with plus disease in zone 1 or posterior zone 2. There appear to be significant advantages to anti-VEGF therapy for this group of patients, including reduced morbidity from treatment and preservation of peripheral retinal function. Close observation for months after injection is critically important, as late recurrence is more likely than after laser, though seldom requiring treatment beyond a second injection.
The BEAT-ROP multicenter study (2011) recently confirmed that primary treatment with a single intravitreal injection of the VEGF inhibitor bevacizumab (Avastin) may be superior to laser for stage 3 ROP with plus disease in zone 1 or posterior zone 2. There appear to be significant advantages to anti-VEGF therapy for this group of patients, including reduced morbidity from treatment and preservation of peripheral retinal function. Close observation for months after injection is critically important, as late recurrence is more likely than after laser, though seldom requiring treatment beyond a second injection.
 Bevacizumab injection may also be beneficial for progressive ERP, after laser or in zone 3, that threatens to cause tractional detachment. Occasional development of TRD following injection in such cases makes some authorities wary of using this approach except in conjunction with surgery to relieve traction.
Bevacizumab injection may also be beneficial for progressive ERP, after laser or in zone 3, that threatens to cause tractional detachment. Occasional development of TRD following injection in such cases makes some authorities wary of using this approach except in conjunction with surgery to relieve traction.
 In the hands of a vitreoretinal surgeon experienced with infant eyes, lens-sparing vitrectomy with or without scleral buckling can prevent progressive deterioration and partially restore vision lost to retinal detachment in stage 4 ROP. The prognosis for eyes with stage 5 ROP remains grim despite recent surgical advances. Eyes that emerge from the acute phase of ROP with significant scarring are at risk for rhegmatogenous retinal detachment from childhood through adulthood.
In the hands of a vitreoretinal surgeon experienced with infant eyes, lens-sparing vitrectomy with or without scleral buckling can prevent progressive deterioration and partially restore vision lost to retinal detachment in stage 4 ROP. The prognosis for eyes with stage 5 ROP remains grim despite recent surgical advances. Eyes that emerge from the acute phase of ROP with significant scarring are at risk for rhegmatogenous retinal detachment from childhood through adulthood.
 Older infants and children with regressed ROP should be followed and treated as necessary for myopia and strabismus, which usually become evident by age 1 to 2 years. Asymmetric cicatricial fundus changes and refractive error increase the risk of amblyopia. Central fixation and binocular fusion may not be achievable in patients with significant macular ectopia from dragging. Children with vision loss from cicatricial changes may need blind services at an early age or low vision care at school age. Lensectomy may improve comfort in eyes with total retinal detachment and glaucoma.
Older infants and children with regressed ROP should be followed and treated as necessary for myopia and strabismus, which usually become evident by age 1 to 2 years. Asymmetric cicatricial fundus changes and refractive error increase the risk of amblyopia. Central fixation and binocular fusion may not be achievable in patients with significant macular ectopia from dragging. Children with vision loss from cicatricial changes may need blind services at an early age or low vision care at school age. Lensectomy may improve comfort in eyes with total retinal detachment and glaucoma.
Differential Diagnosis
 Familial exudative vitreoretinopathy (FEVR) may present at any age including early infancy, often bearing close resemblance to ROP. The condition is autosomal dominant with a wide range of expressions, frequently mild enough to be asymptomatic in parents, but also frequently of sporadic occurrence. Most cases of what appears to be ROP in infants over 2,000 g at birth are probably FEVR.
Familial exudative vitreoretinopathy (FEVR) may present at any age including early infancy, often bearing close resemblance to ROP. The condition is autosomal dominant with a wide range of expressions, frequently mild enough to be asymptomatic in parents, but also frequently of sporadic occurrence. Most cases of what appears to be ROP in infants over 2,000 g at birth are probably FEVR.
 Norrie disease (males only, X-linked recessive) causes a variety of posterior segment changes including vitreous hemorrhage, retinal fold or detachment, and retrolental mass (vitreoretinal dysplasia), which may be present at birth or develop during infancy. Early stages sometimes resemble ROP. Hearing loss and mental deterioration (psychosis or dementia) become evident later in life. Genetic diagnosis is now possible. Abnormality of the Norrie gene (chromosome locus Xp11.3) may play a role in cases of isolated ROP-like retinopathy that develop in larger infants.
Norrie disease (males only, X-linked recessive) causes a variety of posterior segment changes including vitreous hemorrhage, retinal fold or detachment, and retrolental mass (vitreoretinal dysplasia), which may be present at birth or develop during infancy. Early stages sometimes resemble ROP. Hearing loss and mental deterioration (psychosis or dementia) become evident later in life. Genetic diagnosis is now possible. Abnormality of the Norrie gene (chromosome locus Xp11.3) may play a role in cases of isolated ROP-like retinopathy that develop in larger infants.
 Incontinentia pigmenti (Bloch-Sulzberger syndrome) occurs almost exclusively in females, resulting from an X-linked dominant gene usually lethal in males. Retinal vascular development is incomplete at full-term birth. Neovascular lesions in the retinal periphery may closely mimic acute-phase ROP but can develop or progress after infancy. A complex sequence of characteristic skin changes in infancy (culminating in a distinctive pattern of hyperpigmented macules that gradually fade by adulthood) may be overlooked or misinterpreted. Skeletal, dental, and CNS abnormalities are found in some patients (Table 8.13). An affected mother with mild disease may be diagnosed after detection in her child.
Incontinentia pigmenti (Bloch-Sulzberger syndrome) occurs almost exclusively in females, resulting from an X-linked dominant gene usually lethal in males. Retinal vascular development is incomplete at full-term birth. Neovascular lesions in the retinal periphery may closely mimic acute-phase ROP but can develop or progress after infancy. A complex sequence of characteristic skin changes in infancy (culminating in a distinctive pattern of hyperpigmented macules that gradually fade by adulthood) may be overlooked or misinterpreted. Skeletal, dental, and CNS abnormalities are found in some patients (Table 8.13). An affected mother with mild disease may be diagnosed after detection in her child.
References
Flynn JT, Bancalari E, Bachynski BN, et al. Retinopathy of prematurity: diagnosis, severity, and natural history. Ophthalmology 1987;94:620–629.
Reynolds JD, Dobson V, Quinn GE, et al. Evidence-based screening criteria for retinopathy of prematurity: natural history data from the CRYO-ROP and LIGHT-ROP studies. Arch Ophthalmol 2002;120:1470–1476.
Coats Disease
Summary
Coats disease is a form of congenital retinal telangectasia associated with exudative retinal detachment.
Demographics
 Sporadic, more common in boys; unilateral in 90% of cases.
Sporadic, more common in boys; unilateral in 90% of cases.
 Usually detected between ages 3 and 10 years, often on routine vision screening.
Usually detected between ages 3 and 10 years, often on routine vision screening.
 Cases with presentation in infancy tend to be more severe and more often involve girls.
Cases with presentation in infancy tend to be more severe and more often involve girls.
 No significant systemic associations.
No significant systemic associations.
Symptoms and Signs
 Severely decreased vision.
Severely decreased vision.
 May present as leukocoria, or strabismus secondary to decreased vision.
May present as leukocoria, or strabismus secondary to decreased vision.
Ophthalmic Findings
 Telangeictatic retinal vessels with localized aneurysmal dilation, often showing characteristic “light bulb” appearance, usually in the peripheral retina (Figure 8.15A and B). Posterior vessels are often dilated and tortuous.
Telangeictatic retinal vessels with localized aneurysmal dilation, often showing characteristic “light bulb” appearance, usually in the peripheral retina (Figure 8.15A and B). Posterior vessels are often dilated and tortuous.
 Accumulation of yellowish subretinal exudate containing cholesterol crystals may be confined to the macular region (as in Figure 8.15A), or involve the entire retina.
Accumulation of yellowish subretinal exudate containing cholesterol crystals may be confined to the macular region (as in Figure 8.15A), or involve the entire retina.
 Neovascular glaucoma and phthisis bulbi are seen as late complications following total retinal detachment in some cases.
Neovascular glaucoma and phthisis bulbi are seen as late complications following total retinal detachment in some cases.
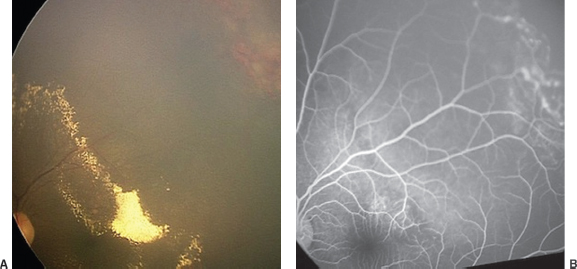
FIGURE 8.15. Coats disease. A: Wide-angle fundus image showing accumulation of hard exudate in the posterior pole as a result of leakage from abnormal vessels seen in retinal periphery. B: Fluorescein angiographic image of same region.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


