RETINAL ANATOMY
Macula and Fovea
The macula histologically contains more than two layers of ganglion cells (Figure 3.1).
Peripheral Retina
 Histological definition: any area with a single layer of ganglion cells.
Histological definition: any area with a single layer of ganglion cells.
 Terminates at the ora serrata located 6 mm posterior to the limbus nasally and 7 mm posterior to the limbus temporally.
Terminates at the ora serrata located 6 mm posterior to the limbus nasally and 7 mm posterior to the limbus temporally.
 Continuous with the nonpigmented epithelium of the pars plana.
Continuous with the nonpigmented epithelium of the pars plana.
Retinal Layers
The layers of the retina are depicted in Figure 3.2.
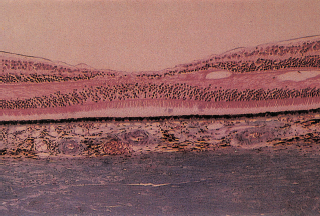
FIGURE 3.1. Histology of a normal macula. Note the multiple nuclei in the ganglion cell layer outside the fovea.
Retinal Circulation
Central retinal artery: supplies the inner two thirds of the retina.
Choriocapillaris: supplies the inner nuclear layer outward to the retinal pigment epithelium (RPE).1
Ophthalmic artery: gives off long and short posterior ciliary arteries.
Long posterior ciliary arteries: supply the choriocapillaris from the optic nerve to the equator.
Short posterior ciliary arteries: supply the optic nerve head and the peripapillary choroid.
Recurrent branches of the anterior ciliary arteries: supply the anterior choriocapillaris.
Vortex veins: one per quadrant; drain the choriocapillaris and form the superior ophthalmic vein that courses through the superior orbital fissure and drains into the cavernous sinus.
Choroid
Four layers:
1. Suprachoroidal space.
2. Stroma.
3. Choriocapillaris.
4. Bruch’s membrane.
Nourishes the outer retina and acts as a heat sink for the retina.
Retinal Pigment Epithelium
 Single-layer hexagonal cuboidal cells of neuroectodermal origin.
Single-layer hexagonal cuboidal cells of neuroectodermal origin.
 Cells in fovea are taller, thinner, and have more abundant, larger melanosomes.
Cells in fovea are taller, thinner, and have more abundant, larger melanosomes.
 Functions of the RPE:
Functions of the RPE:
1. Formation of the outer blood–ocular barrier between the choriocapillaris and the sensory retina via tight junctions. Pump functions to remove subretinal fluid.
2. Phagocytosis of rod and cone outer segments.
3. Vitamin A metabolism.
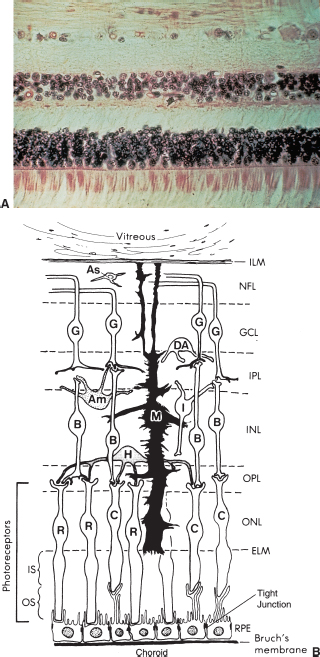
FIGURE 3.2. (A) Histopathology and (B) diagram of the nine retinal layers. The photoreceptors are located at the bottom.
4. Synthesis and degradation of extracellular matrix.
5. Atrophic, hypertrophic, and hyperplastic responses to disease.
6. Light absorption.
7. Heat exchange.
8. Formation of basal lamina.
Bruch Membrane
External to the RPE.
Five layers of Bruch membrane:
1. Basement membrane of the RPE.
2. Inner loose collagenous zone.
3. Middle layer of elastic fibers.
4. Outer loose collagenous zone.
5. Basement membrane of the choriocapillaris.
Photoreceptors
 Outer segments are involved in phototransduction.
Outer segments are involved in phototransduction.
 Inner segments transmit neuronal impulses along axons that synapse with the bipolar and horizontal cells.
Inner segments transmit neuronal impulses along axons that synapse with the bipolar and horizontal cells.
Interneurons, Ganglion Cells, and Glial Cells
See Figure 3.2.
Retinal Layers
1. External limiting membrane: not a true membrane. It is the attachment site of adjacent photoreceptors and Müller cells.
2. Outer plexiform layer: interconnections of photoreceptor synaptic bodies, horizontal, and bipolar cells.
3. Inner nuclear layer: nuclei of bipolar, Müller, horizontal, and amacrine cells.
4. Middle limiting membrane: formed by the attachments of synaptic bodies of photoreceptor cells.
5. Inner plexiform layer: axons of bipolar, amacrine cells, and ganglion cell dendrites and synapses.
6. The nerve fiber layer: ganglion cell axons. The ganglion cell layer contains the ganglion cell bodies.
7. Inner limiting membrane (ILM): not a true membrane; made of footplates of Müller cells.
Ora Serrata
 Boundary between the retina and the pars plana.
Boundary between the retina and the pars plana.
 Retinal blood vessels terminate at the ora serrata, and the vascular supply is the watershed zone between the anterior and the posterior ciliary systems.
Retinal blood vessels terminate at the ora serrata, and the vascular supply is the watershed zone between the anterior and the posterior ciliary systems.
Vitreous
 Four cubic centimeters in volume.
Four cubic centimeters in volume.
 Constitutes four fifths of the entire eye.
Constitutes four fifths of the entire eye.
 Composition is 99% water combined with mucopolysaccharide and hyaluronic acid, which imparts hyperviscosity.
Composition is 99% water combined with mucopolysaccharide and hyaluronic acid, which imparts hyperviscosity.
 Peripheral vitreous attaches to the pars plana, retina, and optic nerve. The strongest attachments are at the vitreous base, optic nerve, and retinal vessels.
Peripheral vitreous attaches to the pars plana, retina, and optic nerve. The strongest attachments are at the vitreous base, optic nerve, and retinal vessels.
References
Apple DJ. Anatomy and histopathology of the macular region.Int Ophthalmol Clin 1981;21:1–9.
Blanks JC. Morphology of the retina. In: Ryan SJ, ed.Retina, 2nd ed., vol. 1. St. Louis, MO: Mosby, 1994:37–54.
Phototransduction
 Four classes of visual pigment:
Four classes of visual pigment:
 Rhodopsin in rods.
Rhodopsin in rods.
 Three pigments in cones.
Three pigments in cones.
 Rods and cones shed their outer segments.
Rods and cones shed their outer segments.
 Retinal rod cells contain many stacked disklike membrane vesicles.
Retinal rod cells contain many stacked disklike membrane vesicles.
 Almost half of the protein in these membranes is a light-absorbing conjugated protein calledrhodopsin.
Almost half of the protein in these membranes is a light-absorbing conjugated protein calledrhodopsin.
 Rhodopsin consists of a protein opsin and tightly bound 11-cis retinal; the aldehyde form of vitamin A.
Rhodopsin consists of a protein opsin and tightly bound 11-cis retinal; the aldehyde form of vitamin A.
 Opsin is synthesized in the inner segments and transported to the base of the outer segment. Opsin is combined with 11-cis retinal to form rhodopsin (Figure 3.3).
Opsin is synthesized in the inner segments and transported to the base of the outer segment. Opsin is combined with 11-cis retinal to form rhodopsin (Figure 3.3).
 The tips of the rod outer segments are shed and phagocytized by the RPE. The rods shed at dawn and the cones shed at dusk. There is a net movement of vitamin A from the outer segments to the RPE during light adaptation, and the reversal of the flow in darkness.
The tips of the rod outer segments are shed and phagocytized by the RPE. The rods shed at dawn and the cones shed at dusk. There is a net movement of vitamin A from the outer segments to the RPE during light adaptation, and the reversal of the flow in darkness.
 Depolarized state: In darkness, there are high levels of cyclic guanosine monophosphate (cGMP) in the outer segments that create open Na+ channels.
Depolarized state: In darkness, there are high levels of cyclic guanosine monophosphate (cGMP) in the outer segments that create open Na+ channels.
 Hyperpolarized state: In the presence of light, there are decreased cGMP levels causing closure of the Na+ channels, which leads to a decrease in neurotransmitter release. Cyclic GMP regulates protein phosphorylation. The bleaching of rhodopsin causes hydrolysis of cGMP through a series of biochemical reactions.
Hyperpolarized state: In the presence of light, there are decreased cGMP levels causing closure of the Na+ channels, which leads to a decrease in neurotransmitter release. Cyclic GMP regulates protein phosphorylation. The bleaching of rhodopsin causes hydrolysis of cGMP through a series of biochemical reactions.
 Light triggers an electrical response in the retina. Presynaptic neurons are depolarized by an action potential (electrical transmission). This leads to an increase in intracellular calcium, which causes a release of neurotransmitters from the synaptic vesicles. Neurotransmitters stimulate postsynaptic receptors. There is then reuptake, catabolism, or diffusion of the neurotransmitter.
Light triggers an electrical response in the retina. Presynaptic neurons are depolarized by an action potential (electrical transmission). This leads to an increase in intracellular calcium, which causes a release of neurotransmitters from the synaptic vesicles. Neurotransmitters stimulate postsynaptic receptors. There is then reuptake, catabolism, or diffusion of the neurotransmitter.
There are three levels of neurotransmission:
1. Light hyperpolarizes the photoreceptors causing a decrease in transmitter release.
2. Bipolar and horizontal cells respond to the decreased neurotransmitter with graded potential.
3. Ganglion cell generates an action potential.
 The neurotransmitters in the retina include the following:
The neurotransmitters in the retina include the following:
Acetylcholine.
Dopamine.
Gamma-aminobutyric acid: inhibits dopaminergic and cholinergic activity in the retina.
Glutamate.
Aspartate (causes excitation block of second order neurons).
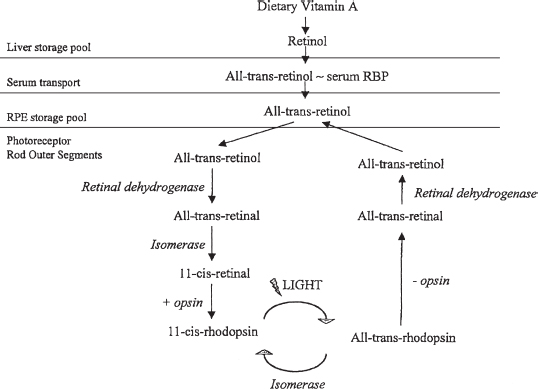
FIGURE 3.3. Biochemical cycle of vitamin A metabolism in the eye. (Adapted fromRetina and vitreous: basic and clinical science course, Section 12, San Francisco, CA: American Academy of Ophthalmology, 1993: Figure 1–2.)
 Energy for visual excitation comes from adenosine triphosphate (ATP) generated from glucose metabolism. There are three pathways of glucose metabolism:
Energy for visual excitation comes from adenosine triphosphate (ATP) generated from glucose metabolism. There are three pathways of glucose metabolism:
1. Glycolysis.
2. Tricarboxylic acid (TCA cycle): generates most of the ATP for the retina.
3. Hexose monophosphate shunt.
Reference
Fundamentals and principles of ophthalmology: retina physiology.Basic and clinical science course. San Francisco, CA: American Academy of Ophthalmology, 1993:179–203.
Electrophysiological Testing
Electroretinogram
See Figure 3.4.
 Evoked by a brief flash of light.
Evoked by a brief flash of light.
 Negative a wave: photoreceptor depolarization.
Negative a wave: photoreceptor depolarization.
 Positive b wave: Müller and bipolar cells.
Positive b wave: Müller and bipolar cells.
 Scotopic electroretinogram (ERG): performed with a blue flash in the dark-adapted state; isolates rod response.
Scotopic electroretinogram (ERG): performed with a blue flash in the dark-adapted state; isolates rod response.
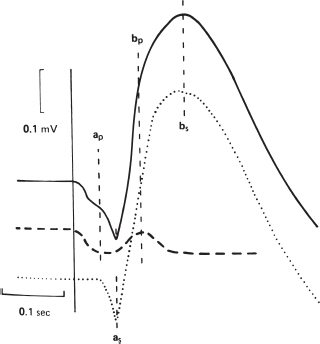
FIGURE 3.4. Analysis of the ERG in a dark-adapted eye (solid line) as the result of photopic (dashed line) and scotopic (dotted line) components. Thea wave is composed of photopic (ap) and scotopic (as) components, and theb wave is similarly composed of photopic (bp) and scotopic (bs) components. (From Miller NR.Walsh and Hoyt’s clinical neuro-ophthalmology, 4th ed., vol. 1. Baltimore, MD: Williams & Wilkins, 1982:36, with permission.)
 Photopic ERG: performed with a bright white flash in the light-adapted state; isolates cone response.
Photopic ERG: performed with a bright white flash in the light-adapted state; isolates cone response.
 Clinical uses:
Clinical uses:
Help diagnose retinitis pigmentosa (RP), pattern dystrophies, and other retinal disorders.
Differentiates ischemic (decreased b:a wave amplitude) from nonischemic central retinal vein occlusion (CRVO).
Assesses retinal toxicity of intraocular foreign body (IOFB).
Multifocal ERG
 Mechanism is based on stimulating macular function while suppressing rod activity with a bright light.
Mechanism is based on stimulating macular function while suppressing rod activity with a bright light.
 Clinical uses include objective assessment of macular function, for example, can be used to detect early hydroxychloroquine toxicity.
Clinical uses include objective assessment of macular function, for example, can be used to detect early hydroxychloroquine toxicity.
Electrooculogram
Electrooculogram (EOG) measures the RPE standing potential.
Arden ratio: maximum light adapted (light peak) to minimum dark adapted (dark trough). Normal value is greater than 1.75.
Clinical uses: dystrophies (i.e., Best disease, which has a normal ERG and an abnormal EOG).
Visual Evoked Potential
Visual evoked potential is the electrical signal generated by the occipital visual cortex.
 Tests macular function.
Tests macular function.
 Clinical uses:
Clinical uses:
Evaluates optic neuropathy, confirms projection of optic nerve fibers in albinism.
Verifies intact visual pathway in preverbal or uncooperative patients.
References
Grand MG, Bressler NM, Brown GC, et al. Retinal physiology and psychophysics. In: Denny M, Taylor F, eds.Basic and clinical science course: retina and vitreous. San Francisco, CA: American Academy of Ophthalmology, 1996:20–40.
Regillo C, Holekamp N, Johnson MW, et al. Retinal physiology and psychophysics. In: Skuta GL, Cantor LB, eds.Basic and clinical science course: retina and vitreous. San Francisco, CA: American Academy of Ophthalmology, 2008–2009:37–38.
Color Vision
Cones have three types of pigment: blue, green, and red.
 Red/green deficiency primarily represents X-linked inheritance.
Red/green deficiency primarily represents X-linked inheritance.
 Blue/yellow deficiencies are usually seen in acquired diseases.
Blue/yellow deficiencies are usually seen in acquired diseases.
 There are two types of color vision tests:
There are two types of color vision tests:
 Pseudoisochromatic plates (Ishihara, Hardy-Rand Littler) on which color numbers stand out from a background of dots. Useful in screening color-deficient individuals but does not classify the deficiency.
Pseudoisochromatic plates (Ishihara, Hardy-Rand Littler) on which color numbers stand out from a background of dots. Useful in screening color-deficient individuals but does not classify the deficiency.
 Panel tests (Farnsworth panel D15, Farnsworth-Munsell 100-hue) discriminate between subtle shades of similar colors:
Panel tests (Farnsworth panel D15, Farnsworth-Munsell 100-hue) discriminate between subtle shades of similar colors:
D15 test involves 15 color tiles that cover the visual spectrum. The subject arranges the tablets in perceived sequence; although the test is not very sensitive, it is fast and accurate.
D15 is useful in retinal diseases because it discriminates congenital from acquired defects.
Reference
Grand MG, Bressler NM, Brown GC, et al. Retinal physiology and psychophysics. In: Denny M, Taylor F, eds.Basic and clinical science course: retina and vitreous. San Francisco, CA: American Academy of Ophthalmology, 1996:20–40.
Retinal Imaging
Intravenous Fluorescein Angiography
 Sodium fluorescein absorbs blue light (465 to 490 nm) and emits green/yellow light (520 to 530 nm).
Sodium fluorescein absorbs blue light (465 to 490 nm) and emits green/yellow light (520 to 530 nm).
 Fluorescein is metabolized by the liver and excreted by the kidneys. One half of usual dose is usually administered in patients with renal failure.
Fluorescein is metabolized by the liver and excreted by the kidneys. One half of usual dose is usually administered in patients with renal failure.
 Pseudofluorescence occurs when nonfluorescent light passes through the entire filter system. The blue excitor filter overlaps into the yellow/green zone, and the yellow/green barrier filter overlaps into the blue zone. This causes nonfluorescent structures to appear fluorescent and can be detected by early photographs before dye injection.
Pseudofluorescence occurs when nonfluorescent light passes through the entire filter system. The blue excitor filter overlaps into the yellow/green zone, and the yellow/green barrier filter overlaps into the blue zone. This causes nonfluorescent structures to appear fluorescent and can be detected by early photographs before dye injection.
 Side effects of fluorescein angiography (FA):
Side effects of fluorescein angiography (FA):
Nausea in 1% to 5% of patients. This usually occurs 30 seconds after injection and lasts approximately 2 to 3 minutes.
Extravasation and local tissue necrosis.
Intra-arterial injection.
Emesis.
Vasovagal reaction.
Allergic reaction.
Discoloration of the skin and urine.
Anaphylaxis.
Nerve palsy.
Neurologic problems.
Thrombophlebitis.
Pyrexia.
Death (extremely rare).
 Normal angiogram choroidal fluorescence (Table 3.1).
Normal angiogram choroidal fluorescence (Table 3.1).
 Two categories of abnormalities (Figure 3.5):
Two categories of abnormalities (Figure 3.5):
1. Hypofluorescence (Figure 3.6).
Blocked fluorescence.
Vascular filling defect.
2. Hyperfluorescence
Pseudofluorescence (seen on red-free photographs before injection of fluorescein).
Optic nerve drusen.
Astrocytic hamartoma.
Sclera.
Exudate.
Scar.
Myelinated nerve fibers.
Foreign body.
Transmitted fluorescence: RPE window defect.
Abnormal vascular fluorescence: tortuosity, dilation, anastomoses, neovascularization (NV), aneurysms, telangiectasis, and tumors.
Leakage: may leak into the vitreous, the disk, the retina, or the choroid.
Table 3.1. Fluorescein angiography phases
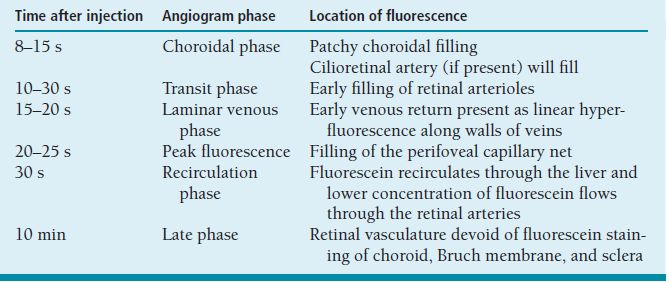
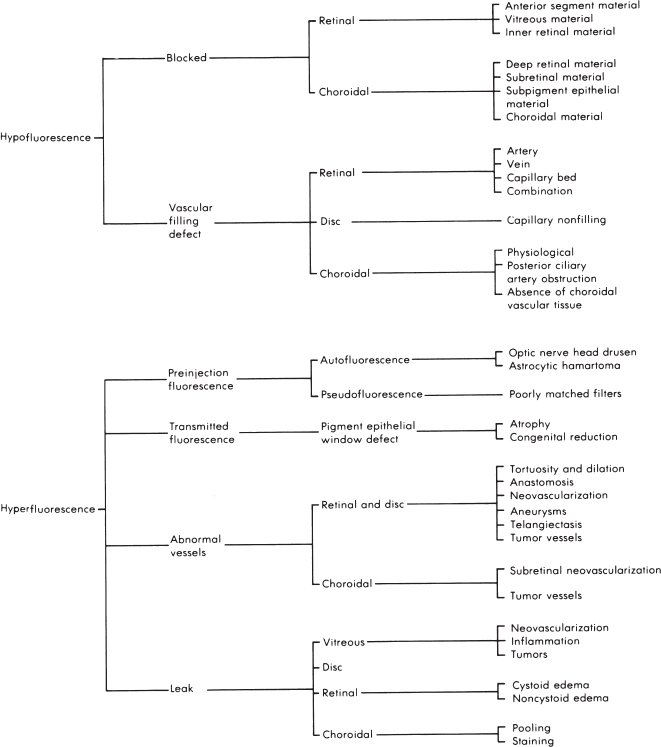
FIGURE 3.5. Flow sheet for abnormal FA.
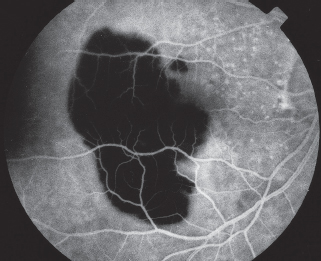
FIGURE 3.6. IVFA demonstrating hypofluorescence due to blockage by a large subretinal hemorrhage. The underlying choroidal fluorescence is obscured by the blood, but the retinal blood vessels are visible anterior to the blood.
Pooling: leakage of fluorescein dye into a distinct anatomic space (i.e., a sensory retinal detachment [RD] such as seen in central serous chorioretinopathy or an RPE detachment).
Staining: leakage of fluorescein diffusely into a tissue as in drusen, scar, or sclera.
Reference
Novotny HR, Alvis DL. A method of photographing fluorescence in circulating blood of the human eye.Am J Ophthalmol 1960;50:176.
Indocyanine Green Video Angiography
 Indocyanine green (ICG) video angiography is a water-soluble tricarbocyanine dye that absorbs and fluoresces in the near-infrared range.
Indocyanine green (ICG) video angiography is a water-soluble tricarbocyanine dye that absorbs and fluoresces in the near-infrared range.
 Less blockage of fluorescence by overlying pigment allowing enhanced imaging of the choroid.
Less blockage of fluorescence by overlying pigment allowing enhanced imaging of the choroid.
 Because ICG dye is highly protein bound (98%), there is minimal escape from the choroidal vessels.
Because ICG dye is highly protein bound (98%), there is minimal escape from the choroidal vessels.
 ICG angiography allows visualization through retinal and subretinal hemorrhages, serous fluid, lipid, and pigment.
ICG angiography allows visualization through retinal and subretinal hemorrhages, serous fluid, lipid, and pigment.
 Contraindications:
Contraindications:
Allergic reactions may occur in patients who have allergies to iodine.
Avoid in liver failure patients, since the dye is metabolized by the liver.
Avoid in pregnant women.
 Clinical uses of ICG angiography:
Clinical uses of ICG angiography:
 Detection of choroidal neovascularization (CNV).
Detection of choroidal neovascularization (CNV).
 Occult CNV with overlying hemorrhage.
Occult CNV with overlying hemorrhage.
 Recurrent CNV.
Recurrent CNV.
 Occult CNV with serous pigment epithelial detachment; detection of classic CNV with an area of occult choroidal neovascular membrane (CNVM) diagnosed by FA.
Occult CNV with serous pigment epithelial detachment; detection of classic CNV with an area of occult choroidal neovascular membrane (CNVM) diagnosed by FA.
 Useful in evaluating intraocular tumors, choroiditis, choroidal vascular disorders, and choroidal infarctions.
Useful in evaluating intraocular tumors, choroiditis, choroidal vascular disorders, and choroidal infarctions.
Reference
Krupsky S, Friedman E, Foster CS, et al. Indocyanine green angiography in choroidal diseases.Invest Ophthalmol Vis Sci 1992;33:723.
Optical Coherence Tomography
 Noninvasive, noncontact and produces cross-sectional images of the retina described as “optical biopsy.” It is based on low-coherence interferometry and uses light waves instead of ultrasound waves used in ultrasound. Reflected light waves from the retina form false-color images that represent the different retinal layers.
Noninvasive, noncontact and produces cross-sectional images of the retina described as “optical biopsy.” It is based on low-coherence interferometry and uses light waves instead of ultrasound waves used in ultrasound. Reflected light waves from the retina form false-color images that represent the different retinal layers.
 Newer generations of spectral domain optical coherence tomography (OCT) are much faster with less motion artifact and show more details than the time-domain OCT scanners.
Newer generations of spectral domain optical coherence tomography (OCT) are much faster with less motion artifact and show more details than the time-domain OCT scanners.
 Clinical uses include differentiating lamellar from full-thickness macular holes, highlighting vitreomacular traction, following treatment responses of cystoid macular edema (CME), neovascular macular degeneration. Spectral domain OCT also delineates external limiting membrane and outer segment-inner segment junctions, which if deficient may explain decreased visual acuity (VA) in many macular diseases (Figure 3.7).
Clinical uses include differentiating lamellar from full-thickness macular holes, highlighting vitreomacular traction, following treatment responses of cystoid macular edema (CME), neovascular macular degeneration. Spectral domain OCT also delineates external limiting membrane and outer segment-inner segment junctions, which if deficient may explain decreased visual acuity (VA) in many macular diseases (Figure 3.7).
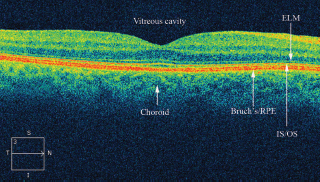
FIGURE 3.7. Normal Cirrus SD-OCT. ELM, external limiting membrane; I, inferior; IS/OS, inner segment outer segment junction; N, nasal; S, superior; T, temporal.
References
Kiernan DF, Mieler WF, Hariprasad SM. Spectral-domain optical coherence tomography: a comparison of modern high-resolution retinal imaging systems.Am J Ophthalmol (Perspectives Invitation) 2010;149(1):18–31.
Kiernan DF, Hariprasad SM, Chin EK, et al. Prospective comparison of high-definition-Cirrus® and Stratus® optical coherence tomography for quantifying retinal thickness.Am J Ophthalmol 2009;147(2):267–275.
Regillo C, Holekamp N, Johnson MW, et al. Retinal physiology and psychophysics. In: Skuta GL, Cantor LB, eds.Basic and clinical science course: retina and vitreous. San Francisco, CA: American Academy of Ophthalmology, 2008–2009: 37–38.
Age-Related Macular Degeneration
Summary
Age-related macular degeneration (ARMD) is the leading cause of significant central visual impairment in the United States in patients over age 50 years. There are two main forms: nonexudative, in which there are no neovascular growths, and exudative, in which NV occurs.
Etiology
Pathogenesis unknown.
Genetic predisposition: allelic variants of genes encoding complement factor H, mutations at chromosome 1q31 at 10q26 and LOC387715 at 10q significantly increase risk of ARMD.
Associations with smoking, hypertension, high cholesterol level, and cardiovascular disease.
Possible nutritional deficiency.
Signs and Symptoms
Blurred vision.
Metamorphopsia.
Central/paracentral scotomas.
Demographics
Predominantly over age 55 years.
Associated with light irides, hyperopia, and female gender.
Nonexudative (nonneovascular) age-related macular degeneration.
Ophthalmic Findings of Nonexudative Age-Related Macular Degeneration
See Figure 3.8.
Drusen (large, soft drusen). By electron microscopy, this material at the level of Bruch membrane is formed of basal laminar and linear deposits. Drusen can be categorized according to size to the following: Small (<64 mm in diameter). Intermediate (64 to 124 μm in diameter). Large (≥125 μm in diameer). Chorioretinal atrophy. RPE hyperpigmentation.
Ophthalmic Findings of Exudative (Neovascular) Age-Related Macular Degeneration
See Figure 3.9.
Subretinal fluid.
Subretinal/vitreous hemorrhage (VH).
Subretinal/intraretinal exudates.
Pigment epithelial detachments.
RPE tears.
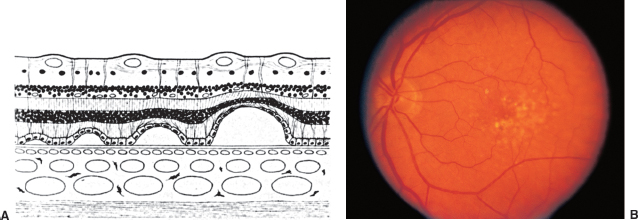
FIGURE 3.8. Age-related macular degeneration. A: Schematic section of retina shows progressively larger detachments of pigment epithelium. Drusen deposit between the pigment epithelium and Bruch membrane. B: Nonexudative age-related macular degeneration with drusen and retinal pigment epithelial changes.
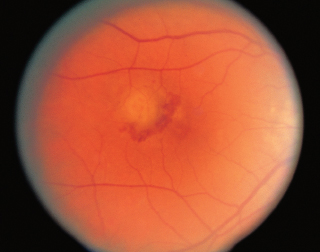
FIGURE 3.9. Exudative age-related macular degeneration with a CNVM surrounded by subretinal hemorrhage.
CNVMs.
Disciform scar.
Systemic Findings
None.
Special Tests
Intravenous fluorescein angiography (IVFA): to determine presence and localization of CNVM for treatment. Two main patterns of CNVM: classic (early bright hyperfluorescence with late leakage) and occult (early stippled hyperfluorescence with late leakage corresponding to fibrovascular pigment epithelium detachments [PEDs], or late leakage of undetermined source).
ICG angiography: may be helpful in evaluating PEDs, occult CNVM.
OCT is currently very useful for follow-up of patients with neovascular ARMD to determine the need of continued treatment and may replace IVFA in many instances (Figure 3.10).
Pathology
Irregular thickening of Bruch membrane leads to cracks through which abnormal neovascular growth from the choriocapillaris may occur.
Disease Course
 A grading scale was developed by the AREDS (Age-Related Eye Disease Study) depending on the presence in each eye of the following:
A grading scale was developed by the AREDS (Age-Related Eye Disease Study) depending on the presence in each eye of the following:
One or more large drusen (1 point).
Pigment abnormalities (1 point).
Bilateral intermediate drusen (1 point).
Neovascular ARMD (2 points).
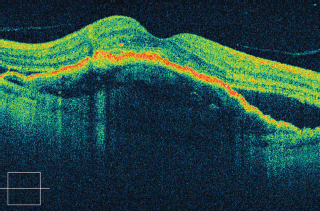
FIGURE 3.10. Cirrus SD-OCT of a patient with neovascular ARMD showing a large pigment epithelial detachment and subretinal fluid.
Points are summed in both eyes to determine the number-related risk of advanced ARMD as follows:
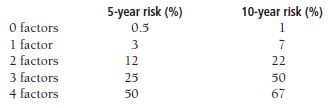
 Severe visual loss associated with advanced ARMD either CNV or geographic atrophy.
Severe visual loss associated with advanced ARMD either CNV or geographic atrophy.
Treatment and Management
Education: Amsler grid testing.
Prophylactic laser photocoagulation was shown not to decrease the incidence of progression to neovascular late ARMD (see study section for the Complications of Age-Related Macular Degeneration Prevention Trial).
The current gold standard treatment are intravitreal antiangiogenesis drugs. Ranibizumab (Lucentis) intravitreal injections have been proven efficacious in FDA registration trials to prevent vision loss and provide visual gain in patients with neovascular ARMD with classic as well as occult CNV (see study section for ANCHOR, MARINA, PIER, and PrONTO studies). Many treatment protocols have been adopted, ranging from monthly injections (MARINA and ANCHOR) to OCT-based guided treatment (PrONTO) to “Treat and Extend.” Bevacizumab (Avastin) intravitreal injections are not FDA approved for use in the eye; however, this treatment modality has been shown in several pilot studies to be beneficial and is currently being tested head to head with ranibizumab in the Comparison of Age-related Macular Degeneration Treatment trial (see study section).
Photodynamic treatment with Verteporfin has also shown some value prior to the advent of antivascular endothelial growth factor (anti-VEGF) therapy in treating neovascular CNV (see study section for the TAP and VIP studies), and it is currently being evaluated for potential combination treatment with anti-VEGF treatment (see study section for the FOCUS and RADICAL studies).
Focal laser photocoagulation according to the MPS study is no longer used for subfoveal or juxtafoveal CNV but may still have a role in some extrafoveal CNV lesions (see study section for MPS study).
Submacular surgery and retinal translocation: may be performed to evacuate acute submacular hemorrhages or rotate the retina away from pathologic RPE/scar. Submacular surgery is not advocated for extracting CNVM associated with ARMD (see study section for the SST trial).
Low-vision aids.
Medications
According to the Age-Related Eye Disease Study I, taking a combination of vitamins (500 mg vitamin C, 400 IU vitamin E, 15 mg beta carotene, 80 mg zinc, and 2 mg cupric oxide to prevent zinc-induced anemia) may decrease the incidence of progression to advanced ARMD by 25% and moderate vision loss by 19% (refer to study section for the AREDS study).
Follow-Up
 If no CNVM, some authors advocate biannual examinations.
If no CNVM, some authors advocate biannual examinations.
 Patients with active CNV are typically followed on a 4- to 6-week basis to determine the need for additional injections.
Patients with active CNV are typically followed on a 4- to 6-week basis to determine the need for additional injections.
Differential Diagnosis
 Idiopathic central serous chorioretinopathy (ICSC) (may have RPE disturbances, serous RDs; seen in young, predominantly male patients).
Idiopathic central serous chorioretinopathy (ICSC) (may have RPE disturbances, serous RDs; seen in young, predominantly male patients).
 Myopic degeneration.
Myopic degeneration.
 Macular dystrophy (e.g., Best disease: has abnormal EOG).
Macular dystrophy (e.g., Best disease: has abnormal EOG).
 RPE pattern dystrophies.
RPE pattern dystrophies.
 Bull’s-eye maculopathies (central macular chorioretinal atrophy seen with various conditions such as chloroquine toxicity, cone dystrophy, fundus flavimaculatus).
Bull’s-eye maculopathies (central macular chorioretinal atrophy seen with various conditions such as chloroquine toxicity, cone dystrophy, fundus flavimaculatus).
 Trauma: choroidal rupture.
Trauma: choroidal rupture.
References
Age-Related Eye Diseases Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E, beta carotene, and zinc for age-related macular degeneration and vision loss. AREDS report 8.Arch Ophthalmol 2001;119:1417–1436.
Ferris FL, Davis MD, Clemons TE, et al. Age-Related Eye Diseases Study (AREDS) Research Group. A simplified severity scale for age-related macular degeneration: AREDS report 18.Arch Ophthalmol 2005;123:1570–1574.
Macular Photocoagulation Study Group. Laser photocoagulation of subfoveal neovascular lesions in age-related macular degeneration: results of a randomized clinical trial.Arch Ophthalmol 1991;109:1219–1231.
Macular Photocoagulation Study Group. Argon laser photocoagulation for neovascular maculopathy after five years: results from randomized clinical trials.Arch Ophthalmol 1991; 109:1109–1114.
Regillo C, Holekamp N, Johnson MW, et al. Retinal physiology and psychophysics. In: Skuta GL, Cantor LB, eds.Basic and clinical science course: retina and vitreous. San Francisco, CA: American Academy of Ophthalmology, 2008–2009:37–38.
Idiopathic Central Serous Chorioretinopathy
Summary
ICSC is a condition characterized by serous elevation of the sensory retina in the macula, typically affecting young male patients.
Etiology
 Pathogenesis unknown; thought to be due to a localized abnormality in the RPE fluid pump.
Pathogenesis unknown; thought to be due to a localized abnormality in the RPE fluid pump.
 May be exacerbated by corticosteroid use.
May be exacerbated by corticosteroid use.
Signs and Symptoms
Blurred vision.
Metamorphopsia.
Micropsia.
Hyperopic shift in refraction (due to elevation of sensory retina).
Demographics
Young to middle-aged adults.
Male-to-female ratio is 8:1 to 10:1.
“Type A” personality.
Ophthalmic Findings
 Serous RD in macula.
Serous RD in macula.
 Subretinal yellowish precipitates.
Subretinal yellowish precipitates.
 Atrophic RPE changes (evidence of previous episodes) in ipsilateral or contralateral eye.
Atrophic RPE changes (evidence of previous episodes) in ipsilateral or contralateral eye.
 Extramacular RPE tracts.
Extramacular RPE tracts.
Systemic Findings
None.
Special Tests
IVFA: classic “smokestack” with focal point of hyperfluorescence that rises and then diffuses laterally in 15% to 20% of cases. Majority have focal point of hyperfluorescence that increases slightly.
OCT shows subretinal fluid and can be used as guide of gradual spontaneous regression and resolution and follow response to treatment (Figure 3.11).
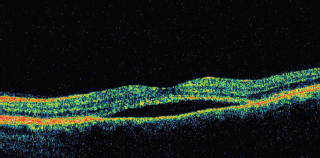
FIGURE 3.11. OCT of a patient with central serous choroidopathy showing subretinal fluid.
Pathology
 Subretinal proteinaceous fluid.
Subretinal proteinaceous fluid.
 Retinal photoreceptors normal unless serous detachment is chronic.
Retinal photoreceptors normal unless serous detachment is chronic.
Disease Course
 Spontaneous resolution of subretinal fluid occurs in 3 to 4 months with improvement of VA to 20/30 or better in over 90% of patients.
Spontaneous resolution of subretinal fluid occurs in 3 to 4 months with improvement of VA to 20/30 or better in over 90% of patients.
 Recurrences may occur in up to 50% of patients.
Recurrences may occur in up to 50% of patients.
 Uncommon complications include CNVM, macular edema, and peripheral chorioretinal atrophic tracts.
Uncommon complications include CNVM, macular edema, and peripheral chorioretinal atrophic tracts.
Treatment and Management
 Observation: Prescribing hyperopic glasses may help to temporize until ICSC resolves.
Observation: Prescribing hyperopic glasses may help to temporize until ICSC resolves.
 Focal laser photocoagulation: may hasten resolution of fluid; however, final VA and recurrence rates are unaffected. Photocoagulation usually is reserved for patients in whom (a) occupational needs require hastened resolution, (b) prolonged leakage persists over 4 to 6 months, or (c) previous episode resulted in a permanent loss of vision.
Focal laser photocoagulation: may hasten resolution of fluid; however, final VA and recurrence rates are unaffected. Photocoagulation usually is reserved for patients in whom (a) occupational needs require hastened resolution, (b) prolonged leakage persists over 4 to 6 months, or (c) previous episode resulted in a permanent loss of vision.
 Photodynamic therapy with Verteporfin using either full fluence or half fluence has shown some promise in some small series in inducing resolution of subretinal fluid with some improvement in VA, but is still experimental.
Photodynamic therapy with Verteporfin using either full fluence or half fluence has shown some promise in some small series in inducing resolution of subretinal fluid with some improvement in VA, but is still experimental.
 Corticosteroids are contraindicated, and in some instances, may exacerbate the condition.
Corticosteroids are contraindicated, and in some instances, may exacerbate the condition.
Medications
None effective.
Differential Diagnosis
 Serous detachments in pregnancy, hypertension, or corticosteroid use: very similar in clinical appearance to ICSC.
Serous detachments in pregnancy, hypertension, or corticosteroid use: very similar in clinical appearance to ICSC.
 Age-related macular degeneration.
Age-related macular degeneration.
 Rhegmatogenous RD: Look for peripheral retinal breaks.
Rhegmatogenous RD: Look for peripheral retinal breaks.
Reference
Gass JDM. Pathogenesis of disciform detachment of the neuroepithelium, II: idiopathic central serous choroidopathy.Am J Ophthalmol 1967;63:587–615.
Cystoid Macular Edema
Summary
CME is the accumulation of fluid in a petalloid pattern in the outer plexiform layer of the macula. It may be seen in many ocular diseases.
Causes of Cystoid Macular Edema
 Postsurgical:
Postsurgical:
Cataract extraction, especially with capsular rupture or vitreous loss.
Vitrectomy.
Cyclophotocoagulation.
Cryopexy.
 Uveitis.
Uveitis.
 Vascular.
Vascular.
Vein occlusion (branch retinal vein occlusion [BRVO], CRVO).
Diabetes mellitus.
 Miscellaneous.
Miscellaneous.
Epiretinal membranes.
RP.
Nicotinic acid maculopathy.
Juvenile X-linked retinoschisis.
Cytomegalovirus (CMV) retinitis.
Etiology
 Mechanism of disease is unknown. Hypotheses include the following:
Mechanism of disease is unknown. Hypotheses include the following:
Inflammation: Perifoveal capillary leakage stimulated by prostaglandins released as a result of inflammation secondary to surgery, uveitis, or other factors.
Vitreous traction: leads to retinal capillary dilation and leakage.
Ultraviolet light: may generate free radicals, leading to prostaglandin release.
Signs and Symptoms
 Unilateral decreased vision or metamorphopsia.
Unilateral decreased vision or metamorphopsia.
 Dulled foveal reflex or foveal cysts noted on slit-lamp biomicroscopy.
Dulled foveal reflex or foveal cysts noted on slit-lamp biomicroscopy.
Demographics
 Depends on etiology.
Depends on etiology.
 Has been reported to occur in a dominantly inherited pattern.
Has been reported to occur in a dominantly inherited pattern.
Ophthalmic Findings
 Foveal cysts or dulled foveal reflex, usually unilateral (can be bilateral if associated with systemic disease).
Foveal cysts or dulled foveal reflex, usually unilateral (can be bilateral if associated with systemic disease).
 Intraocular lens; possible posterior capsule rupture or vitreous strands to wound or iris if associated with complicated cataract extraction.
Intraocular lens; possible posterior capsule rupture or vitreous strands to wound or iris if associated with complicated cataract extraction.
 Hemorrhages, microaneurysms, cotton-wool spots, perimacular edema associated with vascular disease such as diabetic retinopathy, central or BRVO, retinal telangiectasis.
Hemorrhages, microaneurysms, cotton-wool spots, perimacular edema associated with vascular disease such as diabetic retinopathy, central or BRVO, retinal telangiectasis.
 Anterior chamber cell and flare, vitreous cells, other signs of inflammation associated with uveitic causes.
Anterior chamber cell and flare, vitreous cells, other signs of inflammation associated with uveitic causes.
 Pigmentary retinopathy, attenuated retinal vessels, waxy pallor of optic nerve if associated with RP.
Pigmentary retinopathy, attenuated retinal vessels, waxy pallor of optic nerve if associated with RP.
 Distortion of intraretinal vessels, contraction of macular surface secondary to epiretinal fibrosis.
Distortion of intraretinal vessels, contraction of macular surface secondary to epiretinal fibrosis.
Systemic Findings
 Depends on etiology.
Depends on etiology.
 Diabetic patients may have nephropathy, neuropathy, or other microvascular abnormalities.
Diabetic patients may have nephropathy, neuropathy, or other microvascular abnormalities.
 Patients with venous occlusive disease may have signs of systemic vascular disease, hypertension, hypercholesterolemia, etc.
Patients with venous occlusive disease may have signs of systemic vascular disease, hypertension, hypercholesterolemia, etc.
Special Tests
Fluorescein angiographic characteristics:
Focal areas of hyperfluorescence early.
Late pooling of dye in cystoid spaces.
OCT shows the cystic spaces and can be used as a guide to follow the response to treatment (Figure 3.12).
Pathology
Accumulation of edema in outer plexiform layer of macula.
Disease Course
 Depends on etiology.
Depends on etiology.
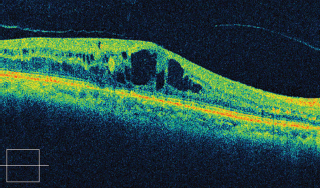
FIGURE 3.12. Cirrus SD-OCT of a patient with CME showing the intraretinal cystic spaces.
 Acute pseudophakic CME may resolve over weeks to months without treatment.
Acute pseudophakic CME may resolve over weeks to months without treatment.
 Chronic pseudophakic CME frequently persists, ultimately resulting in chronic photoreceptor and RPE alterations.
Chronic pseudophakic CME frequently persists, ultimately resulting in chronic photoreceptor and RPE alterations.
 CME associated with diabetic retinopathy gradually progresses and can result in significant visual decline.
CME associated with diabetic retinopathy gradually progresses and can result in significant visual decline.
 CME associated with uveitis waxes and wanes with the underlying uveitis.
CME associated with uveitis waxes and wanes with the underlying uveitis.
Treatment and Management
 Depends on etiology.
Depends on etiology.
 Pseudophakic CME.
Pseudophakic CME.
Most common cause.
Low frequency of occurrence (estimated at 1% to 2% of uncomplicated cataract extractions) has made the completion of a randomized, masked, controlled study difficult.
Stepwise treatment options:
1. Topical nonsteroidal anti-inflammatory drugs (NSAIDs) and/or topical prednisolone for at least 1 month have been shown in several pilot studies to be of benefit.
2. Sub-Tenon corticosteroids (triamcinolone 40 mg/1 mL).
3. Nd: YAG laser vitreolysis or anterior segment reconstruction when indicated.
4. Pars plana vitrectomy.
 Diabetic macular edema: focal or grid laser for clinically significant macular edema (CSME).
Diabetic macular edema: focal or grid laser for clinically significant macular edema (CSME).
 BRVO: focal or grid laser for persistent macular edema and vision 20/40 or worse.
BRVO: focal or grid laser for persistent macular edema and vision 20/40 or worse.
 Uveitis: topical, periocular, and systemic corticosteroids; refractory cases may require immunosuppressives.
Uveitis: topical, periocular, and systemic corticosteroids; refractory cases may require immunosuppressives.
Medications
 Topical NSAIDs (nepafenac, ketorolac, diclofenac, bromfenac) and corticosteroids (prednisolone) are most commonly used for initial treatment of pseudophakic CME. Treatment is instituted at three to four times a day for at least 1 to 2 months and then tapered slowly when vision stabilizes. If topical therapy fails, one may consider sub-Tenon or intravitreal steroid or intravitreal anti-VEGF therapy.
Topical NSAIDs (nepafenac, ketorolac, diclofenac, bromfenac) and corticosteroids (prednisolone) are most commonly used for initial treatment of pseudophakic CME. Treatment is instituted at three to four times a day for at least 1 to 2 months and then tapered slowly when vision stabilizes. If topical therapy fails, one may consider sub-Tenon or intravitreal steroid or intravitreal anti-VEGF therapy.
 Systemic corticosteroids may be beneficial in the treatment of CME associated with uveitis.
Systemic corticosteroids may be beneficial in the treatment of CME associated with uveitis.
 Acetazolamide has shown limited success in the treatment of CME associated with RP and CMV retinitis.
Acetazolamide has shown limited success in the treatment of CME associated with RP and CMV retinitis.
Follow-Up
 Pseudophakic CME: monthly or every other month while on treatment.
Pseudophakic CME: monthly or every other month while on treatment.
 Eight weeks after laser treatment (diabetic and vein occlusion patients).
Eight weeks after laser treatment (diabetic and vein occlusion patients).
Differential Diagnosis
Epiretinal membrane: usually see membrane overlying macula, associated with vessel straightening and tortuosity.
Macular degeneration: Exudative disease can result in CME associated with drusen, subretinal and intraretinal hemorrhage.
Juvenile X-linked retinoschisis, nicotinic retinopathy, and CME associated with RP: These diseases may have the cystoid appearance noted on slit-lamp biomicroscopy but will not leak during angiography.
References
Coscas G, Gaudric A. Natural course of nonaphakic cystoid macular edema.Surv Ophthalmol 1984;28(Suppl):471–484.
Hariprasad SM, Akudman, L, Clever JA, et al. Treatment of cystoid macular edema with the new-generation NSAID Nepafenac 0.1%.Clin Ophthalmol 2009;3(1):147–154.
Hariprasad SM, Callanan D. Topical Nepafenac 0.1% for the treatment of chronic uveitic cystoid macular edema.Retin Cases Brief Rep 2008;2(4):304–308.
Spaide RF, Yannuzzi LA. Cystoid macular edema after cataract surgery.Semin Ophthalmol 1993;8:121–129.
Macular Hole
Summary
A full-thickness round defect in the macula involving all layers from the ILM to the outer segments of the photoreceptor layer.
Etiology (Theories)
 Idiopathic macular holes are caused by tangential traction of the cortical vitreous overlying the fovea.
Idiopathic macular holes are caused by tangential traction of the cortical vitreous overlying the fovea.
 Macular holes may also be seen following trauma, in high myopia, and following chronic CME.
Macular holes may also be seen following trauma, in high myopia, and following chronic CME.
Signs and Symptoms
Blurred vision.
Metamorphopsia.
Demographics
Women more than men.
Sixth decade or older.
Ophthalmic Findings
 Round, punched-out lesion approximately one third disk diameter in size, with a surrounding cuff of subretinal fluid (Table 3.2).
Round, punched-out lesion approximately one third disk diameter in size, with a surrounding cuff of subretinal fluid (Table 3.2).
 Epiretinal fibrosis may be seen at edges, especially prevalent with traumatic holes.
Epiretinal fibrosis may be seen at edges, especially prevalent with traumatic holes.
Systemic Findings
None.
Table 3.2. Classification of idiopathic macular holes

Special Tests
Watzke slit-beam test: The patient notices a break in a thin beam centered over the macular hole.
FA: early hyperfluorescence without late leakage (result of loss of xanthophyll, which is located in the inner layers of the retina).
OCT: differentiates full thickness from lamellar holes and may show vitreous attachment at edges and confirm closure postsurgery and possibly prognosis with intact outer retinal layers at baseline and follow-up (Figure 3.13).
Pathology
Full-thickness loss of retinal tissue from the ILM to the outer segment of the photoreceptor layer. The posterior cortical vitreous may be attached.
Disease Course
 Fifty percent of stage 1 holes may resolve with occurrence of a posterior vitreous detachment (PVD).
Fifty percent of stage 1 holes may resolve with occurrence of a posterior vitreous detachment (PVD).
 Stage 2 holes typically progress to advanced stages.
Stage 2 holes typically progress to advanced stages.
 Progression from stage 1 to fully developed stage 3 or stage 4 hole can occur over a period of weeks or as long as several years (typically within 6 months).
Progression from stage 1 to fully developed stage 3 or stage 4 hole can occur over a period of weeks or as long as several years (typically within 6 months).
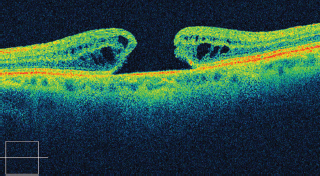
FIGURE 3.13. Cirrus SD-OCT of a patient with full-thickness macular hole.
 VA in patients with full-thickness macular holes is typically stable in the 20/80 to 20/200 range.
VA in patients with full-thickness macular holes is typically stable in the 20/80 to 20/200 range.
 Spontaneous closure of macular holes may occur but is rare. It may be seen with formation of an epiretinal membrane.
Spontaneous closure of macular holes may occur but is rare. It may be seen with formation of an epiretinal membrane.
 Risk of hole development in the normal fellow eye is between 10% and 15%.
Risk of hole development in the normal fellow eye is between 10% and 15%.
Treatment and Management
Intervention consisting of pars plana vitrectomy, stripping of the posterior hyaloid, with and without internal limiting membrane (ILM) peel, and gas tamponade with or without the adjunctive use of autologous serum or other materials can result in closure of the hole and improved vision. Visual improvement of two lines or greater has been achieved in as many as 80% of patients, and dramatic improvement has been reported. The postoperative period requires the patient to maintain a face-down position for periods of 1 to 2 weeks.
Medications
None.
Follow-Up
Without intervention, the course is relatively stable. RD from macular holes is rare and is usually seen in association with high myopia or trauma. If the patient does not desire surgery, annual follow-up is appropriate.
Differential Diagnosis
Epiretinal membrane: Fibrosis overlying macula can result in a pseudohole. May see vessel tortuosity and straightening.
Age-related macular degeneration: central atrophy with surrounding drusen.
CME: edema in petalloid pattern easily discerned on FA.
References
Gass JDM. Idiopathic senile macular hole: its early stages and pathogenesis.Arch Ophthalmol 1988;106:629–639.
Kelly NE, Wendel RT. Vitreous surgery for idiopathic macular hole: results of a pilot study.Arch Ophthalmol 1991;109:654–659.
Epiretinal Membranes
Summary
Epiretinal membranes, also referred to asmacular pucker,surface wrinkling retinopathy, orpreretinal fibrosis, are fibrotic membranes that form by cellular proliferation on the inner surface of the retina.
Etiology
Idiopathic.
Trauma.
Ocular inflammatory disease.
Ocular surgery (especially RD repair and cataract surgery).
Retinal vascular occlusive disease.
Signs and Symptoms
Asymptomatic if mild.
Metamorphopsia.
Blurred vision.
Macropsia.
Rarely diplopia.
Demographics
Most common in elderly (age group in whom PVD most likely to have occurred).
Ophthalmic Findings
 Depends on degree of contraction.
Depends on degree of contraction.
 Classified according to severity.
Classified according to severity.
Cellophane maculopathy: translucent membrane with minimal distortion.
Macular pucker: distinct tissue easily visible on retinal surface with distortion and wrinkling of macular surface.
 PVD present in over 90%.
PVD present in over 90%.
 Simple membranes may be visible as a mild sheen to the retina with irregular light reflex.
Simple membranes may be visible as a mild sheen to the retina with irregular light reflex.
 May be thickened and opaque.
May be thickened and opaque.
 Distortion of retinal vessels, with tortuosity and straightening.
Distortion of retinal vessels, with tortuosity and straightening.
 Foveal ectopia.
Foveal ectopia.
 Retinal striae.
Retinal striae.
 Pseudohole of macula.
Pseudohole of macula.
 Various degrees of macular edema.
Various degrees of macular edema.
 Traction elevation of macula in severe cases.
Traction elevation of macula in severe cases.
Systemic Findings
None.
Special Tests
Fluorescein angiographic characteristics:
Vascular tortuosity and straightening.
Retinal vascular leakage if contraction significant.
OCT can identify the membrane, associated macular edema, and used to follow progression and response to vitrectomy (Figure 3.14).
Pathology
 May be composed of different cell types, including retinal pigment epithelial cells, fibrocytes, fibrous astrocytes, inflammatory cells, and macrophages.
May be composed of different cell types, including retinal pigment epithelial cells, fibrocytes, fibrous astrocytes, inflammatory cells, and macrophages.
 Interlacing network of cells and collagen adherent to ILM.
Interlacing network of cells and collagen adherent to ILM.
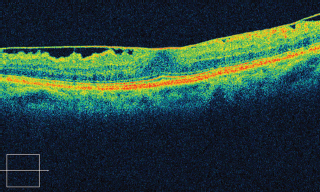
FIGURE 3.14. Cirrus SD-OCT showing epiretinal membrane and loss of foveal depression.
Disease Course
 Vision ranges from normal to worse than 20/200 (<5%).
Vision ranges from normal to worse than 20/200 (<5%).
 Most patients, 20/70 or better.
Most patients, 20/70 or better.
 Vision usually stable once membrane formed.
Vision usually stable once membrane formed.
Treatment and Management
 Observation if vision minimally affected. Look carefully for retinal holes or tears on examination.
Observation if vision minimally affected. Look carefully for retinal holes or tears on examination.
 If significant visual impairment, vitrectomy and membrane stripping should be considered.
If significant visual impairment, vitrectomy and membrane stripping should be considered.
Medications
None.
Follow-Up
Annual examination unless vision worsens.
Differential Diagnosis
Retinal vascular disease: BRVO or retinal telangiectasis can produce edema and vascular abnormalities; differentiated by FA.
Age-related macular degeneration: subretinal fibrosis versus preretinal fibrosis.
References
De Bustros S, Thompson JT, Michels RG, et al. Vitrectomy for idiopathic epiretinal membranes causing macular pucker.Ophthalmology 1988;72:692–695.
Smiddy WE, Maguire AM, Green WR, et al. Idiopathic epiretinal membranes: ultrastructural characteristics and clinicopathologic correlation.Ophthalmology 1989;96:811–821.
Pathologic Myopia
Summary
Pathologic myopia is progressive degeneration associated with myopia of −6.0 diopters or greater and excess axial elongation.
Etiology
Presumed multifactorial (e.g., genetic, environmental).
Signs and Symptoms
Blurred vision.
Scotoma.
Metamorphopsia.
Photopsias.
Floaters.
Demographics
Most common in Asians, least common in blacks.
Women more frequently than men.
Associated with higher education.
Ophthalmic Findings
 Chorioretinal atrophy, particularly in posterior pole (Figure 3.15).
Chorioretinal atrophy, particularly in posterior pole (Figure 3.15).
 Peripapillary crescent, tilted disks.
Peripapillary crescent, tilted disks.
 Lacquer cracks: breaks in Bruch membrane.
Lacquer cracks: breaks in Bruch membrane.
 Subretinal hemorrhages: from breaks in Bruch membrane.
Subretinal hemorrhages: from breaks in Bruch membrane.
 CNVM, Forster-Fuchs spot.
CNVM, Forster-Fuchs spot.
 Lattice degeneration, retinal tears, RD.
Lattice degeneration, retinal tears, RD.
 Vitreous detachment, syneresis.
Vitreous detachment, syneresis.
 Posterior staphyloma.
Posterior staphyloma.
 Strabismus, anisometropic amblyopia.
Strabismus, anisometropic amblyopia.
 Glaucoma.
Glaucoma.
A staphyloma represents an area of ectatic sclera with absent or severely atrophic overlying choroidal or retinal tissue.
Systemic Findings
None.
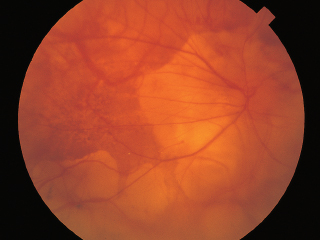
FIGURE 3.15. Multiple areas of chorioretinal atrophy due to myopic degeneration.
Special Tests
 IVFA: to determine presence of CNVM.
IVFA: to determine presence of CNVM.
 A-scan ultrasonography to measure axial length.
A-scan ultrasonography to measure axial length.
Pathology
 Posterior staphyloma lined with atrophic choroid.
Posterior staphyloma lined with atrophic choroid.
 Chorioretinal atrophy of posterior pole.
Chorioretinal atrophy of posterior pole.
 RPE hyperpigmentation/hypopigmentation.
RPE hyperpigmentation/hypopigmentation.
 Vitreous syneresis.
Vitreous syneresis.
Disease Course
Neovascular complications tend to occur during adulthood. Some patients who develop CNVM may have spontaneous regression of the NV.
Treatment
 Laser photocoagulation of extrafoveal CNVM. Complications of laser include progressive RPE atrophy around treated area. Photodynamic therapy (PDT) with Verteporfin has shown promise in subfoveal CNV secondary to pathologic myopia (see study section, VIP study). Some pilot studies have also shown promise of anti-VEGF intravitreal injections.
Laser photocoagulation of extrafoveal CNVM. Complications of laser include progressive RPE atrophy around treated area. Photodynamic therapy (PDT) with Verteporfin has shown promise in subfoveal CNV secondary to pathologic myopia (see study section, VIP study). Some pilot studies have also shown promise of anti-VEGF intravitreal injections.
 Laser photocoagulation of retinal tears.
Laser photocoagulation of retinal tears.
 Scleral buckling for RD.
Scleral buckling for RD.
 Vitrectomy for posterior retinal breaks and RD.
Vitrectomy for posterior retinal breaks and RD.
Medications
None.
Follow-Up
Patients should monitor their central vision with Amsler grids. Patients should be educated on the symptoms of RD.
Differential Diagnosis
Choroideremia.
ARMD: See p. 106.
Presumed ocular histoplasmosis syndrome (POHS): See p. 250.
References
Curtin BJ, Karlin DB. Axial length measurements and fundus changes of the myopic eye.Am J Ophthalmol 1971;71:42–53.
Jalkh AE, Weiter JJ, Trempe CL, et al. Choroidal neovascularization in degenerative myopia: role of laser photocoagulation.Ophthalmic Surg 1987;18:721–725.
Angioid Streaks
Summary
Angioid streaks are breaks in Bruch membrane, usually radiating from the optic disk. Fifty percent are associated with systemic disorders.
Etiology
Unknown; associations with elastic tissue diseases.
Signs and Symptoms
Asymptomatic.
Blurred vision.
Scotoma.
Metamorphopsia.
Demographics (PEPSI)
Pseudoxanthoma elasticum (PXE).
Ehlers-Danlos syndrome.
Paget disease.
Sickle cell disease association.
Idiopathic.
Ophthalmic Findings
RPE changes radiating from optic disk (reddish brown or gray color) (Figure 3.16).
Nearly always bilateral.
CNVM.
Subretinal hemorrhage: may occur with minor trauma.
Macular degeneration.
“Peau d’orange” changes: with PXE; diffuse RPE mottling.
Optic disk drusen: may be seen with PXE.
Systemic Findings
PXE: elastic tissue disease causing “plucked-chicken skin,” gastrointestinal (GI) tract bleeding, cardiac disease.
Paget disease: progressive connective tissue disease causing increased bony mass, high alkaline phosphatase.
Hemoglobinopathies (e.g., sickle cell disease, thalassemias). Complications such as CNVM rare with angioid streaks associated with this group of disorders.
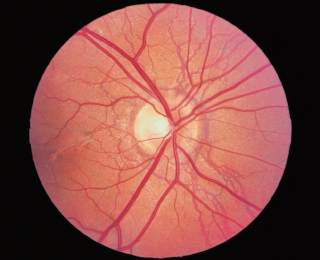
FIGURE 3.16. Angioid streaks radiating from the optic nerve.
Ehler-Danlos syndrome: connective tissue disorder, hyperextensible skin.
Systemic Associations of Angioid Streaks
 Pseudoxanthoma elasticum
Pseudoxanthoma elasticum
 Ehlers-Danlos syndrome Paget disease
Ehlers-Danlos syndrome Paget disease
 Sickle cell and other hemoglobinopathies
Sickle cell and other hemoglobinopathies
 Idopathic
Idopathic
Special Tests
IVFA shows hyperfluorescence of streaks; helps show CNVM.
Pathology
 Thickened Bruch membrane (basophilia and calcification).
Thickened Bruch membrane (basophilia and calcification).
 Elastic degeneration of Bruch membrane.
Elastic degeneration of Bruch membrane.
Disease Course
Patients may initially be asymptomatic but lose vision over time.
Treatment
Medical, dermatologic consult.
Avoid trauma.
Laser photocoagulation for CNVM: has a high recurrence rate. PDT and anti-VEGF off-label treatment may be of some benefit but have not been tested prospectively.
Medications
None.
Follow-Up
Education and Amsler grid testing.
Differential Diagnosis
 ARMD.
ARMD.
 Myopic degeneration: lacquer cracks.
Myopic degeneration: lacquer cracks.
 Presumed ocular histoplasmosis: Ohio-Mississippi River Valley, peripapillary pigment ring, peripheral punched-out chorioretinal scars.
Presumed ocular histoplasmosis: Ohio-Mississippi River Valley, peripapillary pigment ring, peripheral punched-out chorioretinal scars.
 Choroidal rupture: history of trauma, crescent-shaped lesions radial to optic disk.
Choroidal rupture: history of trauma, crescent-shaped lesions radial to optic disk.
Reference
Clarkson JG, Altman RD. Angioid streaks.Surv Ophthalmol 1982;26:235–246.
Photic Retinopathy
Summary
Degeneration of retinal photoreceptors may occur from photochemical injury.
Etiology
 Sun gazing. May occur with direct or indirect viewing of the sun. May follow viewing of the sun after observing an eclipse.
Sun gazing. May occur with direct or indirect viewing of the sun. May follow viewing of the sun after observing an eclipse.
 Ophthalmic instruments (e.g., operating microscope, fiberoptic endoilluminator). Prolonged operating time and intensity of light beam are risk factors.
Ophthalmic instruments (e.g., operating microscope, fiberoptic endoilluminator). Prolonged operating time and intensity of light beam are risk factors.
 Arc welding without wearing protective eyewear.
Arc welding without wearing protective eyewear.
Signs and Symptoms
Decreased vision within several hours after exposure.
Central/paracentral scotomas.
Headache.
Metamorphopsia.
Solar Retinopathy
Demographics
Sungazers.
Psychosis: drug-induced or psychiatric illness.
Ophthalmic Findings
VA 20/40 to 20/100 or worse, short term.
Usually bilateral.
Yellow spot in fovea that becomes reddish several days later.
RPE changes.
Lamellar hole.
Systemic Findings
None.
Special Tests
IVFA: Initially normal. May have central staining. Weeks later, may have only mild RPE defects.
OCT: Abnormal reflectivity in the outer foveal retina, fragmentation or interruption of the inner high reflective layer.
Pathology
Photoreceptor destruction thought due to free radical formation and tissue oxidation, RPE necrosis.
Disease Course
 VA may spontaneously improve.
VA may spontaneously improve.
 May have residual central/paracentral scotoma.
May have residual central/paracentral scotoma.
Treatment
Observation for spontaneous improvement.
Medications
None.
Follow-Up
Education to prevent sun gazing, use of protective eyewear.
Light Toxicity From Ophthalmic Instruments
Demographics
 Patients who have undergone recent cataract surgery (high-intensity coaxial light from surgical microscope).
Patients who have undergone recent cataract surgery (high-intensity coaxial light from surgical microscope).
 Vitreoretinal surgical patients.
Vitreoretinal surgical patients.
Ophthalmic Findings
 Round or oval retinal lesions, usually in parafoveal location.
Round or oval retinal lesions, usually in parafoveal location.
 Lesions usually within arcade vessels.
Lesions usually within arcade vessels.
 RPE mottling and atrophy weeks after surgery.
RPE mottling and atrophy weeks after surgery.
Systemic Findings
None.
Special Tests
IVFA: staining of acute lesion. Later in course, IVFA shows window defects in region of involvement.
Disease Course
 Vision may return to normal several months after surgery.
Vision may return to normal several months after surgery.
 May have persistent paracentral scotoma.
May have persistent paracentral scotoma.
Treatment
Observation.
Medications
None.
Follow-Up
Minimize intraocular surgery times and direct light exposure (e.g., use of pupil occluders during surgery).
Differential Diagnosis
CNVM.
Drug toxicity.
References
Green WR, Robertson DM. Pathologic findings of photic retinopathy in the human eye.Am J Ophthalmol 1991;112:520–527.
Jorge R, Costa RA, Quirino LS, et al. OCT findings in patients with late solar retinopathy.Am J Ophthalmol 2004;137: 1139–1143.
Tso MOM, Woodford BJ. Effect of photic injury on the retinal tissues.Ophthalmology 1983;90:952–963.
Drug Toxicity
Phenothiazines
Summary
Phenothiazine use can result in ocular toxicity, manifest by various degrees of pigmentary retinopathy. This has been most commonly seen with thioridazine (Mellaril) and chlorpromazine (Thorazine).
Etiology
The exact cause of toxicity is unknown. Phenothiazines are absorbed by melanin, resulting in concentration in uveal tissues and RPE.
Acute retinopathy can be seen 3 to 8 weeks after thioridazine use in doses of more than 800 mg/day.
Chlorpromazine retinopathy is typically milder. It is usually seen after high doses for prolonged periods (2,400 mg/day for 1 year).
Signs and Symptoms
Blurred vision.
Dyschromatopsia.
Nyctalopia.
Ophthalmic Findings
Pigmentary retinopathy.
Confluent areas of RPE depigmentation.
Abnormal pigmentation of eyelids, conjunctiva, cornea, and lens capsule.
Systemic Findings
Psychotic disorders.
Manic-depressive illness.
Special Tests
 Visual-field abnormalities.
Visual-field abnormalities.
 Abnormal dark adaptation.
Abnormal dark adaptation.
 IVFA may reveal a wide spectrum of RPE abnormalities ranging from mild alterations to extensive areas of RPE and choriocapillaris atrophy.
IVFA may reveal a wide spectrum of RPE abnormalities ranging from mild alterations to extensive areas of RPE and choriocapillaris atrophy.
 ERG: ranges from normal (early toxicity) to attenuated (severe toxicity).
ERG: ranges from normal (early toxicity) to attenuated (severe toxicity).
Pathology
 Atrophy of the RPE and choriocapillaris.
Atrophy of the RPE and choriocapillaris.
 Atrophy and disorganization of the photoreceptor outer segments.
Atrophy and disorganization of the photoreceptor outer segments.
Disease Course
 Early toxicity: visual complaints associated with a normal fundus picture or a fine pigmentary retinopathy.
Early toxicity: visual complaints associated with a normal fundus picture or a fine pigmentary retinopathy.
 Immediate cessation of medication may result in reversal of visual and fundus abnormalities.
Immediate cessation of medication may result in reversal of visual and fundus abnormalities.
 Late toxicity: Continued use of medication can lead to widespread RPE and choriocapillaris atrophy, which may progress despite cessation of medication.
Late toxicity: Continued use of medication can lead to widespread RPE and choriocapillaris atrophy, which may progress despite cessation of medication.
 Recovery of vision may occur slowly over time.
Recovery of vision may occur slowly over time.
Treatment and Management
Immediate discontinuation of phenothiazine (retinopathy may still progress).
Medications
None.
Follow-Up
Periodic examinations to document recovery or progression after cessation.
Differential Diagnosis
RP: extensive RPE atrophy associated with attenuated vessels, optic nerve pallor.
Cancer-associated retinopathy: widespread RPE atrophy associated with underlying malignancy.
Reference
Miller FS III, Bunt-Milam AH, Kalina RE. Clinical-ultrastructural study of thioridazine retinopathy.Ophthalmology 1982;89: 1478–1488.
Tamoxifen
Summary
Tamoxifen, an antiestrogen that has been found to be an effective therapeutic agent in breast cancer, can cause a crystalline retinopathy, macular edema, and visual loss.
Etiology
 The exact mechanism of toxicity is unknown.
The exact mechanism of toxicity is unknown.
 Initial reports of toxicity were in patients who received high doses of tamoxifen (total doses > 90 g); these doses are no longer prescribed.
Initial reports of toxicity were in patients who received high doses of tamoxifen (total doses > 90 g); these doses are no longer prescribed.
 Low-dosage (tamoxifen 10 mg b.i.d.) long-term therapy may cause toxicity.
Low-dosage (tamoxifen 10 mg b.i.d.) long-term therapy may cause toxicity.
Signs and Symptoms
Blurred vision.
Metamorphopsia.
Diplopia.
Demographics
 Typically in women with history of breast cancer.
Typically in women with history of breast cancer.
 Rarely seen in men undergoing hormonal therapy.
Rarely seen in men undergoing hormonal therapy.
Ophthalmic Findings
Bilateral whorl-like corneal opacities.
Refractile crystals at the level of the inner retina (Figure 3.17).
Macular edema.
RPE abnormalities.
Optic neuritis.
Systemic Findings
History of breast cancer.
Special Tests
Fluorescein angiographic characteristics:
 Pinpoint macular lesions that hyperfluoresce early and leak late.
Pinpoint macular lesions that hyperfluoresce early and leak late.
 Crystalline lesions are hyperfluorescent.
Crystalline lesions are hyperfluorescent.
OCT:
Foveolar cystoids space, loss of photoreceptors, and lack of macular thickening.
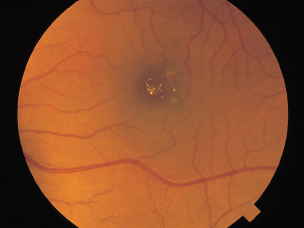
FIGURE 3.17. A 53-year-old woman with multiple crystalline deposits in inner retina after 8 years of tamoxifen treatment.
Pathology
 Refractile lesions in the nerve fiber and inner plexiform layers.
Refractile lesions in the nerve fiber and inner plexiform layers.
 May represent products of axonal degeneration.
May represent products of axonal degeneration.
Disease Course
 Toxicity usually not seen in patients receiving low-dose therapy unless long course of treatment (>7 years) with large cumulative dose (>10 g).
Toxicity usually not seen in patients receiving low-dose therapy unless long course of treatment (>7 years) with large cumulative dose (>10 g).
 Toxicity in patients receiving low-dose therapy is slowly progressive.
Toxicity in patients receiving low-dose therapy is slowly progressive.
Treatment and Management
 Referral to oncologist for consideration of change of therapy if signs of toxicity.
Referral to oncologist for consideration of change of therapy if signs of toxicity.
 Toxicity from high-dose treatment remains stable after discontinuation of drug.
Toxicity from high-dose treatment remains stable after discontinuation of drug.
 Toxicity from low-dose treatment has shown regression of retinopathy with visual recovery after drug discontinuation.
Toxicity from low-dose treatment has shown regression of retinopathy with visual recovery after drug discontinuation.
Medications
None effective.
Follow-Up
Other crystalline maculopathies:
Canthaxanthine maculopathy: skin-tanning agent that can cause a doughnut-like pattern of superficial retinal crystals in the superficial retina.
Oxalosis: Primary or secondary oxalosis may cause a crystalline retinopathy. Diagnosis is aided by detection of urinary oxalate.
Bietti crystalline dystrophy: tapetoretinal degeneration associated with posterior pole crystals. Begins in third decade, autosomal recessive inheritance.
Autosomal dominant crystalline dystrophy: crystalline retinopathy seen predominantly in young female patients.
Calcified macular drusen: Refractile lesions are frequently accompanied by noncalcified drusen.
Talc retinopathy: refractile crystals in the retina seen in intravenous (i.v.) drug abusers who inject crushed oral medications containing talc compounds.
References
Gualino V, Cohen SY, Delyfer MN, et al. OCT findings of in tamoxifen retinopathy.Am J Ophthalmol 2005;140: 757–758.
Heier JS, Dragoo RA, Enzenauer RW, et al. Screening for ocular toxicity in asymptomatic patients treated with tamoxifen.Am J Ophthalmol 1994;117:772–775.
Kaiser-Kupfer MI, Kupfer C, Rodrigues MM. Tamoxifen retinopathy: a clinicopathologic report.Ophthalmology 1981;88:89–93.
Chloroquine and Hydroxychloroquine
Summary
Prolonged use of chloroquine and hydroxychloroquine (Plaquenil), agents used for the treatment of rheumatoid arthritis, amebiasis, malaria, and systemic lupus erythematosus can cause degeneration of the RPE and sensory retina.
Etiology
 Mechanism of retinopathy not known.
Mechanism of retinopathy not known.
 Toxicity seen with chronic use of chloroquine in doses greater than 250 mg/day, or hydroxychloroquine in doses greater than 6.5 mg/kg/day.
Toxicity seen with chronic use of chloroquine in doses greater than 250 mg/day, or hydroxychloroquine in doses greater than 6.5 mg/kg/day.
 Hydroxychloroquine is much less toxic to the eye.
Hydroxychloroquine is much less toxic to the eye.
Signs and Symptoms
Visual loss.
Paracentral scotoma.
Demographics
Toxicity from hydroxychloroquine is most likely in elderly, underweight patients (the toxicity is dose and duration dependent).
Ophthalmic Findings
Corneal deposits.
Bull’s-eye maculopathy: Figure 3.18.
Generalized retinal pigmentary degeneration.
Retinal vessel attenuation.
Optic disk pallor.
Systemic Findings
Rheumatoid arthritis.
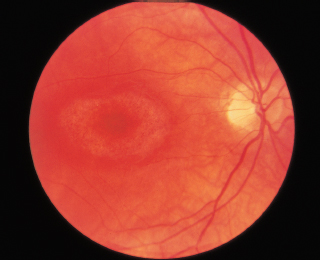
FIGURE 3.18. RPE depigmentation in bull’s-eye configuration resulting from chloroquine retinopathy.
Systemic lupus erythematosus.
Amebiasis.
Malaria.
Special Tests
Amsler grid: central and paracentral abnormalities, red Amsler may be more sensitive.
Humphrey 10-2 visual field examination with a red test object: detects abnormalities in central 20 degrees.
IVFA: bull’s-eye pattern of hyperfluorescence.
Pathology
 Chloroquine is concentrated in the RPE.
Chloroquine is concentrated in the RPE.
 RPE depigmentation, rod and cone receptor loss occurs in the macula.
RPE depigmentation, rod and cone receptor loss occurs in the macula.
Disease Course
 Early toxicity is evident in paracentral scotomas.
Early toxicity is evident in paracentral scotomas.
 Central scotoma develops with increasing toxicity.
Central scotoma develops with increasing toxicity.
 Early visual loss may be reversible with immediate cessation of treatment.
Early visual loss may be reversible with immediate cessation of treatment.
 If central visual loss and/or absolute scotoma is present, progressive loss may occur despite discontinuation of medication.
If central visual loss and/or absolute scotoma is present, progressive loss may occur despite discontinuation of medication.
Treatment and Management
If toxicity detected, discontinue drug immediately.
Medications
None.
Follow-Up
Chloroquine toxicity is rare today, since the large majority of patients previously given this medication have been converted to regimen of hydroxychloroquine.
Patients without underlying macular disease or evidence of toxicity should be given an Amsler grid for self-monitoring and can be followed annually.
Patients with underlying macular disease (i.e., macular degeneration) or taking more than 6.5 mg/kg/day of hydroxychloroquine should be followed more frequently.
Differential Diagnosis
Cone dystrophy: bull’s-eye maculopathy similar, but central vision affected earlier, and to a greater degree. ERG would reveal greater involvement of photopic response.
Stargardt disease: bull’s-eye maculopathy with retinal flecks. Florescein angiography reveals silent choroid.
ARMD: can occasionally see bilateral macular RPE atrophy, associated with surrounding drusen (see p. 106).
Benign concentric annular dystrophy: bull’s-eye maculopathy in young patients with minimal central visual loss.
References
Easterbrook M. The ocular safety of hydroxychloroquine.Semin Arthritis Rheum 1993;23:62–67.
Weiner A, Sandberg MA, Gaudio AR, et al. Hydroxychloroquine retinopathy.Am J Ophthalmol 1991;112:528–534.
Methoxyflurane
Summary
Use of the nonflammable anesthetic methoxyflurane can result in secondary oxalosis being evident in the eye by crystalline deposits in the posterior pole and midperiphery.
Etiology
Prolonged general anesthesia with the inhalational anesthetic methoxyflurane, especially in the presence of underlying renal dysfunction.
Signs and Symptoms
Normal VA.
Ophthalmic Findings
 Numerous yellow-white punctate crystalline deposits scattered about the posterior pole and midperiphery.
Numerous yellow-white punctate crystalline deposits scattered about the posterior pole and midperiphery.
 Deposits may course along the retinal arteries.
Deposits may course along the retinal arteries.
Systemic Findings
Renal dysfunction (may occur before or as a result of methoxyflurane anesthesia).
Pathology
 Birefringent crystalline deposits in RPE.
Birefringent crystalline deposits in RPE.
 Crystalline deposits composed of calcium oxalate.
Crystalline deposits composed of calcium oxalate.
Medications
None effective.
Differential Diagnosis
Canthaxanthine maculopathy: skin-tanning agent that can cause a doughnut-like pattern of superficial retinal crystals in the superficial retina.
Bietti crystalline dystrophy: tapetoretinal degeneration associated with posterior pole crystals. Begins in third decade, autosomal recessive inheritance.
Autosomal dominant crystalline dystrophy: crystalline retinopathy seen predominantly in young female patients.
Calcified macular drusen: refractile lesions are frequently accompanied by noncalcified drusen, other changes of macular degeneration.
Talc retinopathy: refractile crystals in the retina seen in i.v. drug abusers who inject crushed oral medications containing talc compounds.
Tamoxifen retinopathy: bilateral crystalline retinopathy seen in breast cancer patients undergoing hormonal therapy.
References
Bullock JD, Albert DM. Flecked retina: appearance secondary to oxalate crystals from methoxyflurane anesthesia.Arch Ophthalmol 1975;93:26–30.
Novak MA, Roth AS, Levine MR. Calcium oxalate retinopathy associated with methoxyflurane anesthesia.Retina 1988;8:230–236.
Canthaxanthine
Summary
Canthaxanthine is an oral tanning agent that may cause a bilateral crystalline retinopathy.
Etiology
May develop in patients taking canthaxanthine regularly over an extended period.
Signs and Symptoms
VA usually normal.
Ophthalmic Findings
 Symmetric doughnut-like pattern of retinal crystals in the macula.
Symmetric doughnut-like pattern of retinal crystals in the macula.
 Crystals can be found in the superficial retinal layers.
Crystals can be found in the superficial retinal layers.
 Patients may develop corneal crystals.
Patients may develop corneal crystals.
Systemic Findings
None.
Special Tests
Fluorescein angiographic characteristics:
Typically normal.
May show a bull’s-eye pattern of hyperfluorescence.
Pathology
 Lipid-soluble crystals in inner retinal layers and ciliary body.
Lipid-soluble crystals in inner retinal layers and ciliary body.
 Crystals probably represent a canthaxanthine-lipoprotein complex.
Crystals probably represent a canthaxanthine-lipoprotein complex.
Disease Course
 Toxicity is dose dependent.
Toxicity is dose dependent.
 Usually a dose of 19 g or greater is necessary to induce retinopathy.
Usually a dose of 19 g or greater is necessary to induce retinopathy.
 Retinopathy may be seen at lower doses if underlying RPE disease, ocular hypertension, beta carotene use.
Retinopathy may be seen at lower doses if underlying RPE disease, ocular hypertension, beta carotene use.
Treatment and Management
 Crystals may gradually disappear after agent is discontinued.
Crystals may gradually disappear after agent is discontinued.
 May take 1 year or more for crystals to disappear.
May take 1 year or more for crystals to disappear.
Medications
None effective.
Differential Diagnosis
Tamoxifen retinopathy: bilateral crystalline retinopathy seen in breast cancer patients undergoing hormonal therapy.
Oxalosis: Primary or secondary oxalosis may cause a crystalline retinopathy. Diagnosis is aided by detection of urinary oxalate.
Bietti crystalline dystrophy: tapetoretinal degeneration associated with posterior pole crystals. Begins in third decade, autosomal recessive inheritance.
Autosomal dominant crystalline dystrophy: crystalline retinopathy seen predominantly in young female patients.
Calcified macular drusen: refractile lesions are frequently accompanied by noncalcified drusen.
Talc retinopathy: refractile crystals in the retina seen in i.v. drug abusers who inject crushed oral medications containing talc compounds.
References
Daicker B, Schiedt K, Adnet JJ, et al. Canthaxanthin retinopathy: an investigation by light and electron microscopy and physicochemical analysis.Graefes Arch Clin Exp Ophthalmol 1987;225:189–197.
Harnois C, Samson J, Malenfant M, et al. Canthaxanthin retinopathy: anatomic and functional reversibility.Arch Ophthalmol 1989;107:538–540.
Diabetic Retinopathy
Summary
Diabetic retinopathy correlates with pathogenetically underlying systemic diabetes mellitus.
Etiology (Theories)
 Glycosylation of tissue proteins causes cell damage.
Glycosylation of tissue proteins causes cell damage.
 The polyol pathway (aldose reductase) results in an accumulation of intracellular sorbitol, which causes basement membrane thickening and damages pericytes.
The polyol pathway (aldose reductase) results in an accumulation of intracellular sorbitol, which causes basement membrane thickening and damages pericytes.
Signs and Symptoms
Blurring, distortion of vision.
Decreased night vision.
Floaters.
Decreased color vision.
Demographics
 Leading cause of blindness in ages 20 to 64 years.
Leading cause of blindness in ages 20 to 64 years.
 Twenty-five percent of diabetics have diabetic retinopathy.
Twenty-five percent of diabetics have diabetic retinopathy.
Ophthalmic Findings of Nonproliferative Diabetic Retinopathy
See Figure 3.19.
Microaneurysms.
Dilated capillaries.
Dot-blot nerve-fiber layer hemorrhages.
Hard exudates.
Retinal edema.
Cotton-wool spots.
Systemic Findings
Diabetic nephropathy.
Polyneuropathy.
Hypertension found in 22% of type I and 58% of type II diabetics.
Special Tests
 IVFA identifies macular capillary nonperfusion, macular edema, subtle areas of NV, or capillary dropout.
IVFA identifies macular capillary nonperfusion, macular edema, subtle areas of NV, or capillary dropout.
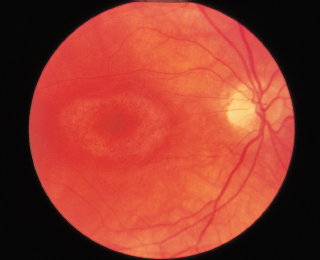
FIGURE 3.19. NPDR with hard exudates and microaneurysms.
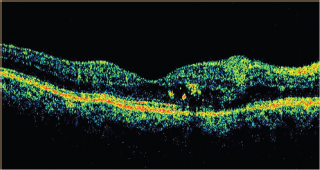
FIGURE 3.20. OCT of a patient with diabetic macular edema showing diffuse intraretinal edema and intraretinal lipid exudates.
 Ultrasonography identifies tractional retinal detachment (TRD) in eyes with opaque media.
Ultrasonography identifies tractional retinal detachment (TRD) in eyes with opaque media.
 Color fundus photography.
Color fundus photography.
OCT identifies macula edema, measures retinal thickness, and is reflective of the macula (Figure 3.20).
Pathology
 Thickening of the vascular basement membrane.
Thickening of the vascular basement membrane.
 Decreased number of pericytes relative to the number of endothelial cells.
Decreased number of pericytes relative to the number of endothelial cells.
Disease Course and Natural Progression
Incidence of retinopathy increases with the duration of diabetes and patient age (Table 3.3).
Treatment and Management
CSME id defined as one or more of the following features(figure 3.21):
 1.Thickening at or within 500 γm of the foveal avascular zone (FAZ).
1.Thickening at or within 500 γm of the foveal avascular zone (FAZ).
 2.Hard exudate at or within 500 γm of the FAZ with associated thickening of the adjacent retina.
2.Hard exudate at or within 500 γm of the FAZ with associated thickening of the adjacent retina.
 3.A zone of retinal thickening 1 disk area or larger,any part of which is within 1 disk diameter of the center of the macula.
3.A zone of retinal thickening 1 disk area or larger,any part of which is within 1 disk diameter of the center of the macula.
 CSME: According to the Early Treatment for Diabetic Retinopathy Study (ETDRS), focal and/or grid laser is beneficial for these eyes.
CSME: According to the Early Treatment for Diabetic Retinopathy Study (ETDRS), focal and/or grid laser is beneficial for these eyes.
Intravitreal Anti-VEGF therapy.
Table 3.3. Incidence of diabetic retinopathy

 NPDR.
NPDR.
Mild: occasional microaneurysms.
Moderate: more microaneurysms and scattered hard exudates or cotton-wool spots.
Severe: the presence of one of the following:
Four quadrants of severe retinal hemorrhages.
Two quadrants of venous beading.
One quadrant of moderately severe IRMA. If two of these are present, this is termedvery severe NPDR. Most patients are advised to wait until high-risk characteristics (HRCs) of proliferative diabetic retinopathy (PDR) are reached to initiate panretinal photocoagulation (PRP). If follow-up is unreliable, PRP may be initiated sooner.
Medications
Intensive insulin therapy can delay the onset of and slow the progression of diabetic retinopathy, nephropathy, and neuropathy in type I diabetics. See Diabetes Control and Complications Trial (DCCT).
Follow-Up
Differential Diagnosis
 Hypertensive retinopathy (arteriovenous [AV] nicking, copper wiring, elevated blood pressure).
Hypertensive retinopathy (arteriovenous [AV] nicking, copper wiring, elevated blood pressure).
 Collagen vascular disease (anti-ssA, anti-ssB, ANA, rheumatoid factor, c-ANCA, arthritis, dermatologic findings, lupus erythematous, polyarteritis nodosa, Wegener granulomatosis).
Collagen vascular disease (anti-ssA, anti-ssB, ANA, rheumatoid factor, c-ANCA, arthritis, dermatologic findings, lupus erythematous, polyarteritis nodosa, Wegener granulomatosis).
 Acquired immunodeficiency syndrome (AIDS) retinopathy (human immunodeficiency virus [HIV]–positive serology).
Acquired immunodeficiency syndrome (AIDS) retinopathy (human immunodeficiency virus [HIV]–positive serology).
 Cardiac embolic disease (Hollenhorst, calcific plaques, talc).
Cardiac embolic disease (Hollenhorst, calcific plaques, talc).
 Sickle cell retinopathy (positive sickle cell preparation, hemoglobin electrophoresis, peripheral nonperfusion, sea-fan NV, black sunburst).
Sickle cell retinopathy (positive sickle cell preparation, hemoglobin electrophoresis, peripheral nonperfusion, sea-fan NV, black sunburst).
 Radiation retinopathy (history of radiation therapy).
Radiation retinopathy (history of radiation therapy).
Table 3.4. Guidelines for examinations for diabetic retinopathy

Table 3.5. Follow up guidelines for diabetic eye diseases

 Vasculitis (sarcoidosis, toxoplasmosis, Eales disease, systemic lupus erythematosus, syphilis, vascular sheathing present).
Vasculitis (sarcoidosis, toxoplasmosis, Eales disease, systemic lupus erythematosus, syphilis, vascular sheathing present).
 Leukemia (history, peripheral blood smear, bone marrow biopsy, Roth spots).
Leukemia (history, peripheral blood smear, bone marrow biopsy, Roth spots).
References
Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus.N Engl J Med 1993;329:977–986.
Early Treatment Diabetic Retinopathy Study Research Group. Photocoagulation for Diabetic Macular Edema: ETDRS Report 1.Arch Ophthalmol 1985;103:1796–1806.
Proliferative Diabetic Retinopathy
Summary
The presence of newly formed blood vessels or fibrous tissue arising from the retina or optic disk and extending along the inner surface of the retina or disk or into the vitreous cavity.
Etiology
Closure of retinal arterioles causes nonperfusion and ischemia and stimulates the release of vasoproliferative factors stimulating NV from the retina, optic nerve, or iris.
Signs and Symptoms
Blurred vision.
Distortion.
Floaters.
Demographics
 Twenty-six percent of patients who have had diabetes for 25 to 50 years develop PDR.
Twenty-six percent of patients who have had diabetes for 25 to 50 years develop PDR.
 Type I diabetics have a higher risk of developing PDR than type II patients.
Type I diabetics have a higher risk of developing PDR than type II patients.
 There are 65,000 new cases of PDR per year and 8,000 new cases of blindness due to diabetic retinopathy per year in the United States.
There are 65,000 new cases of PDR per year and 8,000 new cases of blindness due to diabetic retinopathy per year in the United States.
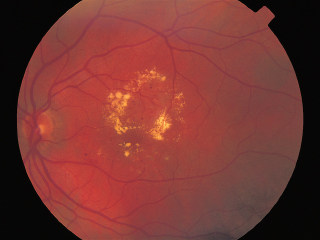
FIGURE 3.21. CSME with circinate ring of hard exudates and retinal thickening involving the fovea.
Ophthalmic Findings
Neovascularization of the disk (NVD) or elsewhere (NVE) (Figure 3.22).
Vitreous or preretinal hemorrhages.
Preretinal fibrosis.
TRD.
Systemic Findings
Nephropathy.
Polyneuropathy.
Hypertension.
Carotid occlusive disease.
Special Tests
IVFA: hyperfluorescence and leakage of dye from neovascular vessels (Figure 3.23).
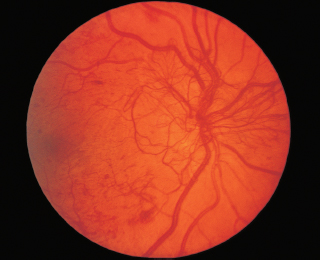
FIGURE 3.22. PDR with extensive NVD.
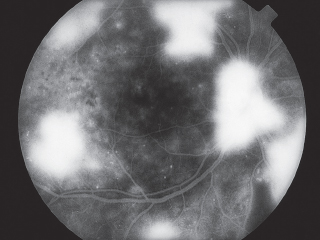
FIGURE 3.23. IVFA demonstrating multiple areas of hyperfluorescence due to leakage from neovascularization in PDR.
Ultrasonography: in cases with opaque media to evaluate underlying TRD.
Color fundus photography: helpful in following the regression or the progression of NV.
OCT: useful in following areas of macular hemorrhage or macula traction due to ERM formation.
Pathology
Growth of new vessels on retinal surface along posterior hyaloid.
Disease Course and Natural Progression
 Continued new vessel growth on the retinal surface in a cycle of proliferation and regression.
Continued new vessel growth on the retinal surface in a cycle of proliferation and regression.
 Proliferation of fibrous tissue accompanies vascular growth.
Proliferation of fibrous tissue accompanies vascular growth.
 Regressed vessels appear sheathed.
Regressed vessels appear sheathed.
 A partial PVD and contraction of fibrous tissue pulls neovascular vessels and creates a VH. As the partial PVD spreads in a posterior to anterior direction, it cannot be released from areas of adhesion between the vitreous and the fibrovascular proliferation.
A partial PVD and contraction of fibrous tissue pulls neovascular vessels and creates a VH. As the partial PVD spreads in a posterior to anterior direction, it cannot be released from areas of adhesion between the vitreous and the fibrovascular proliferation.
 Fibrovascular contraction also leads to traction retinal detachment and macular dragging (Figure 3.24).
Fibrovascular contraction also leads to traction retinal detachment and macular dragging (Figure 3.24).
 Occasionally, vitreous separation is completed and there is involution of the abnormal vessels.
Occasionally, vitreous separation is completed and there is involution of the abnormal vessels.
Treatment and Management
 For severe NPDR and PDR without HRCs, careful follow-up with prompt PRP if HRCs develop.
For severe NPDR and PDR without HRCs, careful follow-up with prompt PRP if HRCs develop.
 Full-scatter PRP, which includes 1,200 to 1,600 laser burns, 500-mm, 0.1-second argon burns in two sessions, is recommended for patients with HRCs.
Full-scatter PRP, which includes 1,200 to 1,600 laser burns, 500-mm, 0.1-second argon burns in two sessions, is recommended for patients with HRCs.
 Intravitreal Anti-VEGF treatment.
Intravitreal Anti-VEGF treatment.
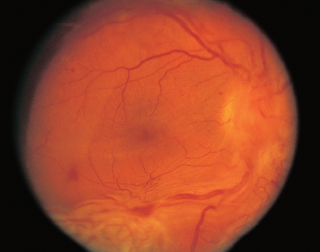
FIGURE 3.24. Fibrovascular proliferation causing TRD along inferotemporal arcade.
HRCs defined by the Diabetic Retinopathy Study:
 1. NVD,at leat one third of disk are.
1. NVD,at leat one third of disk are.
 2. NVD,on or with 1 disk diameter of the disk of the and vitreous or preretinal hemorrhage.
2. NVD,on or with 1 disk diameter of the disk of the and vitreous or preretinal hemorrhage.
 3. NVD,at least halt of disk area and vitreous or prerentinal hemorhage.
3. NVD,at least halt of disk area and vitreous or prerentinal hemorhage.
panretinal photocoagulation:

 1. power: 600 to 1,600 mW, increase power as needed to achieve moderate retinal burn.
1. power: 600 to 1,600 mW, increase power as needed to achieve moderate retinal burn.

 Spot size: 500 mm.
Spot size: 500 mm.

 Duration:0.1 second.
Duration:0.1 second.

 comments:Divide into two sessions. Start outside of arcades, place burn one burn width apart.
comments:Divide into two sessions. Start outside of arcades, place burn one burn width apart.

 Recommended for patients who achieve HRCs.
Recommended for patients who achieve HRCs.
 2. NVD,on or with 1 disk diameter of the disk of the and vitreous or preretinal hemorrhage.
2. NVD,on or with 1 disk diameter of the disk of the and vitreous or preretinal hemorrhage.
 3. NVD,at least halt of disk area and vitreous or prerentinal hemorhage.
3. NVD,at least halt of disk area and vitreous or prerentinal hemorhage.
Medical Therapy
 Intensive insulin therapy may delay and slow the progression of diabetic retinopathy (DCCT); however, patients have a higher incidence of hypoglycemic episodes.
Intensive insulin therapy may delay and slow the progression of diabetic retinopathy (DCCT); however, patients have a higher incidence of hypoglycemic episodes.
 Aspirin treatment is not contraindicated for management of other concomitant systemic diseases (ETDRS).
Aspirin treatment is not contraindicated for management of other concomitant systemic diseases (ETDRS).
 Follow-up recommended every month after treatment until vessels regress.
Follow-up recommended every month after treatment until vessels regress.
 Supplemental PRP is recommended for eyes with nonregressing NV, increasing NV, new VH, or new areas of NV.
Supplemental PRP is recommended for eyes with nonregressing NV, increasing NV, new VH, or new areas of NV.
Surgical Therapy
 Results of the Diabetic Vitrectomy Study showed that eyes of type I diabetics with nonclearing VH of greater than 3 months duration benefit from early vitrectomy. However, with the advent of modern-day vitrectomy technology, intervention is typically sooner.
Results of the Diabetic Vitrectomy Study showed that eyes of type I diabetics with nonclearing VH of greater than 3 months duration benefit from early vitrectomy. However, with the advent of modern-day vitrectomy technology, intervention is typically sooner.
 Other indications for vitrectomy are bilateral VH and TRD involving the macula.
Other indications for vitrectomy are bilateral VH and TRD involving the macula.
Differential Diagnosis
Retinopathy of prematurity (ROP; history of prematurity, prolonged oxygenation, low birth weight, peripheral NV, macular dragging).
Sickle cell retinopathy (sea-fan NV, positive sickle cell preparation, hemoglobin electrophoresis).
Sarcoidosis (gallium scan, angiotensin-converting enzyme [ACE], abnormal chest x-ray [CXR], vasculitis, vitreous cells).
CRVO (unilateral, diffuse flame-shaped hemorrhages, venous dilation, tortuosity).
BRVO (hemorrhages involving one sector of the retina).
References
Diabetic Retinopathy Study Research Group. Indications For Photocoagulation Treatment of Diabetic Retinopathy: DRS Report 14.Int Ophthalmol Clin 1987;26:239–253.
Diabetic Retinopathy Vitrectomy Study Research Group. Early vitrectomy for severe vitreous hemorrhage in diabetic retinopathy: two-year results of a randomized trial—DRVS Report 2.Arch Ophthalmol 1985;103:1644–1652.
Radiation Retinopathy
Summary
Radiation retinopathy is characterized by a delayed-onset postradiation progressive occlusive vasculopathy that leads to capillary nonperfusion, large-vessel occlusion, retinal vascular incompetence, and retinal NV.
Etiology
Radiation-induced damage to endothelial cells of retinal vasculature.
Signs and Symptoms
Asymptomatic.
Decreased vision from macular ischemia or VHs.
Demographics
 The incidence depends on total dose and daily fraction size.
The incidence depends on total dose and daily fraction size.
 Most studies report 30 to 35 Gy needed to produce retinopathy.
Most studies report 30 to 35 Gy needed to produce retinopathy.
 Cobalt plaque:
Cobalt plaque:
 A mean of 150 Gy causes foveal damage.
A mean of 150 Gy causes foveal damage.
 Retinopathy occurs 4 to 32 months after treatment.
Retinopathy occurs 4 to 32 months after treatment.
 External beam radiation:
External beam radiation:
 A mean of 49 Gy causes foveal damage.
A mean of 49 Gy causes foveal damage.
 Retinopathy occurs 7 to 36 months after treatment.
Retinopathy occurs 7 to 36 months after treatment.
 Proton-beam radiation: A mean of 70 Gy leads to foveal damage.
Proton-beam radiation: A mean of 70 Gy leads to foveal damage.
Ophthalmic Findings
 Nerve fiber layer infarction (cotton-wool spots), retinal hemorrhages, microaneurysms, telangiectasis, perivascular sheathing, exudation.
Nerve fiber layer infarction (cotton-wool spots), retinal hemorrhages, microaneurysms, telangiectasis, perivascular sheathing, exudation.
 Late changes include RPE atrophy, central retinal artery occlusion (CRAO), CRVO, and NV.
Late changes include RPE atrophy, central retinal artery occlusion (CRAO), CRVO, and NV.
Systemic Findings
None.
Special Tests
IVFA: capillary nonperfusion, microaneurysms, hyperfluorescence, telangiectasis, NV, cotton-wool spots, hard exudates, retinal hemorrhages, CME, sheathing, perivasculitis, optic disk edema, disk NV.
OCT: useful in following macular edema.
Pathology
 Focal loss of capillary endothelial cells and pericytes leading to occlusive vascular disease.
Focal loss of capillary endothelial cells and pericytes leading to occlusive vascular disease.
 Subsequent loss of ganglion cells, cystic changes in the outer plexiform and inner nuclear layers, and thickening of the vessel walls with preferential damage to the inner retinal layers.
Subsequent loss of ganglion cells, cystic changes in the outer plexiform and inner nuclear layers, and thickening of the vessel walls with preferential damage to the inner retinal layers.
 Choroidal infarction may also be seen and demonstrated on ICG angiography.
Choroidal infarction may also be seen and demonstrated on ICG angiography.
Disease Course and Natural Progression
 Widespread capillary closure and ischemia lead to retinal and disk NV and proliferation of fibrous tissue.
Widespread capillary closure and ischemia lead to retinal and disk NV and proliferation of fibrous tissue.
 Contraction of the fibroglial tissue leads to VH and TRD.
Contraction of the fibroglial tissue leads to VH and TRD.
 Anterior segment NV can lead to neovascular glaucoma.
Anterior segment NV can lead to neovascular glaucoma.
Treatment and Management
 Focal laser photocoagulation for macular edema.
Focal laser photocoagulation for macular edema.
 PRP for retinal ischemia and NV.
PRP for retinal ischemia and NV.
 Hyperbaric oxygen has been attempted, but effect is short lived and depends on continued treatment.
Hyperbaric oxygen has been attempted, but effect is short lived and depends on continued treatment.
 Intravitreal Anti-VEGF therapy to assist in Neovascular regression.
Intravitreal Anti-VEGF therapy to assist in Neovascular regression.
Medications
None.
Follow-Up
Every 2 to 4 months.
Differential Diagnosis
 Diabetic retinopathy (diabetes; hemorrhages; microaneurysms more prominent that cotton-wool spots).
Diabetic retinopathy (diabetes; hemorrhages; microaneurysms more prominent that cotton-wool spots).
 Multiple branch retinal artery occlusions (BRAOs) (whitening along distribution of branch retinal artery; may see emboli; hemorrhages uncommon).
Multiple branch retinal artery occlusions (BRAOs) (whitening along distribution of branch retinal artery; may see emboli; hemorrhages uncommon).
 Multiple venous occlusions (more hemorrhages; dilated tortuous veins).
Multiple venous occlusions (more hemorrhages; dilated tortuous veins).
 Ocular ischemic syndrome (midperipheral hemorrhages; low central retinal artery perfusion pressure; dilated veins).
Ocular ischemic syndrome (midperipheral hemorrhages; low central retinal artery perfusion pressure; dilated veins).
References
Brown GC, Shields JA, Sanburn G, et al. Radiation retinopathy.Ophthalmology 1982;89:1494–1501.
Stollard HB. Radiant energy as a pathogenic and a therapeutic agent in ophthalmic disorders.Br J Ophthalmol 1933;N126(Suppl 6): 100–126.
Sickle Cell Retinopathy
Summary
Sickle cell retinopathy is characterized by retinal hemorrhages, nonperfusion, and NV as a result of arteriolar and capillary occlusion.
Etiology
Intravascular sickling, hemolysis, hemostasis, and thrombosis cause peripheral arteriolar occlusion, which leads to capillary nonperfusion and eventually to NV.
Signs and Symptoms
Asymptomatic.
Decreased vision.
Floaters secondary to VH.
Demographics
 Hemoglobin AS (HbAS) affects 8% of black Americans.
Hemoglobin AS (HbAS) affects 8% of black Americans.
 Hemoglobin SS (HbSS) affects 0.4% of black Americans (produces the most severe systemic sickling disease).
Hemoglobin SS (HbSS) affects 0.4% of black Americans (produces the most severe systemic sickling disease).
 Hemoglobin SC (HbSC) affects 0.2% of black Americans (most common to have retinopathy).
Hemoglobin SC (HbSC) affects 0.2% of black Americans (most common to have retinopathy).
 Hemoglobin AC (HbAC) affects 2%.
Hemoglobin AC (HbAC) affects 2%.
 Thalassemia (HbS-Thal) affects 0.3%.
Thalassemia (HbS-Thal) affects 0.3%.
Ophthalmic Findings
See Figure 3.25.
Salmon patch hemorrhages (intraretinal hemorrhages).
Refractile deposits (resorbed hemorrhages).
Black sunbursts (RPE hypertrophy).
Sea-fan NV.
Systemic Findings
Chronic anemia.
Aseptic necrosis of the head of the femur.
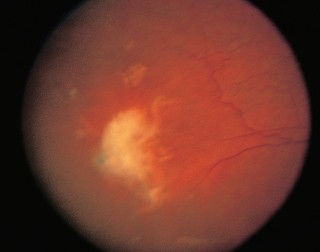
FIGURE 3.25. Sickle cell retinopathy with peripheral neovascularization.
Cerebrovascular accidents (CVAs).
Abdominal infarctions.
Bacterial infections (Salmonella organisms).
HbAS, HbAC: asymptomatic.
HbSC, HbS-Thal: mild anemia.
Special Tests
Sickle cell preparation.
Hemoglobin electrophoresis.
IVFA: nonperfusion, AV anastomoses at the border of perfused and nonperfused retina, sea-fan NV (Figure 3.26).
OCT: Macular Edema
Disease Course and Natural Progression
Stage I: Peripheral arterial occlusion.
Stage II: Peripheral AV anastomoses.
Stage III: Sea-fan NV.
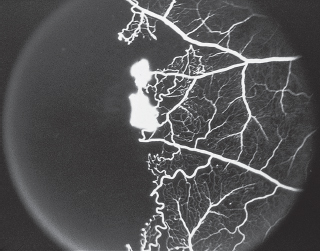
FIGURE 3.26. IVFA demonstrating peripheral nonperfusion capillary dropout and neovascularization.
Stage IV: VH.
Stage V: TRD.
Treatment and Management
 For proliferative sickle retinopathy, PRP, cryotherapy, and diathermy are available treatment modalities. Avoid treating the feeder vessel directly, since this may result in VH.
For proliferative sickle retinopathy, PRP, cryotherapy, and diathermy are available treatment modalities. Avoid treating the feeder vessel directly, since this may result in VH.
 Vitrectomy is recommended for nonclearing VH, rhegmatogenous, traction, or combined RDs.
Vitrectomy is recommended for nonclearing VH, rhegmatogenous, traction, or combined RDs.
Medications
None. Avoid carbonic anhydrase inhibitors, which can worsen sickling.
Follow-Up
Every 3 to 4 months.
Differential Diagnosis
 Eales disease (male patients, unilateral or bilateral peripheral vasculitis).
Eales disease (male patients, unilateral or bilateral peripheral vasculitis).
 PDR (macular ischemia, edema, posterior nonperfusion, NV along major arcades).
PDR (macular ischemia, edema, posterior nonperfusion, NV along major arcades).
 BRVO (intraretinal hemorrhages along a vessel in one sector of the retina).
BRVO (intraretinal hemorrhages along a vessel in one sector of the retina).
 BRAO (arteriole embolus, retinal edema, retinal infarction).
BRAO (arteriole embolus, retinal edema, retinal infarction).
 ROP (prematurity, low birth weight, oxygen exposure, macular dragging).
ROP (prematurity, low birth weight, oxygen exposure, macular dragging).
 Familiar exudative vitreoretinopathy (FEVR) (autosomal dominant inheritance, macular dragging): hyperviscosity (leukemia, lymphoma, macroglobulinemia, polycythemia, multiple myeloma, Roth spots, serous RDs, cotton-wool spots).
Familiar exudative vitreoretinopathy (FEVR) (autosomal dominant inheritance, macular dragging): hyperviscosity (leukemia, lymphoma, macroglobulinemia, polycythemia, multiple myeloma, Roth spots, serous RDs, cotton-wool spots).
 Sarcoidosis (elevated ACE, abnormal CXR, abnormal gallium scan, vascular sheathing, vitreous cells, granulomatous uveitis).
Sarcoidosis (elevated ACE, abnormal CXR, abnormal gallium scan, vascular sheathing, vitreous cells, granulomatous uveitis).
 Ocular ischemic syndrome (painful visual loss, anterior chamber cells, midperipheral microaneurysms and hemorrhages, carotid stenosis, low retinal artery perfusion pressure).
Ocular ischemic syndrome (painful visual loss, anterior chamber cells, midperipheral microaneurysms and hemorrhages, carotid stenosis, low retinal artery perfusion pressure).
References
Cohen SB, Fletcher ME, Goldberg MS, et al. Diagnosis and management of ocular complications of sickle hemoglobinopathies, I–V.Ophthalmic Surg 1986;17:57–59, 110–116, 184–188, 312–315, 369–374.
Goldberg MS. Classification and pathogenesis of proliferative sickle retinopathy.Am J Ophthalmol 1971;71:649–665.
Hypertensive Retinopathy
Summary
Hypertensive retinopathy is a retinal vascular condition secondary to systemic hypertension. The signs range from arterial narrowing to disk swelling and retinal hemorrhages.
Etiology
Elevation of systemic blood pressure causes focal and generalized constriction of the retinal arterioles mediated by autoregulation.
Signs and Symptoms
Usually asymptomatic.
Demographics
Fifty million Americans have hypertension.
Ophthalmic Findings
See Figure 3.27.
Focal or generalized constriction of retinal arterioles.
Cotton-wool spots.
Intraretinal lipid.
Intraretinal hemorrhages.
BRAO.
BRVO.
CRVO.
Retinal arterial macroaneurysms.
Microaneurysms.
Venous congestion.
Elschnig spots and Siegrist streaks (from nonperfusion of the choriocapillaris with ischemia of overlying RPE and outer retina).
Systemic Findings
Patients may have renal failure.
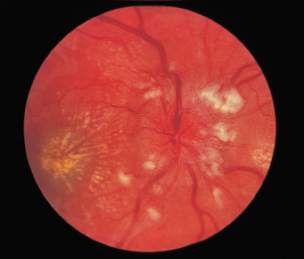
FIGURE 3.27. Malignant hypertension with disk edema, macular star, and multiple cotton-wool spots.
Myocardial infarction.
CVAs related to systemic hypertension.
Special Tests
IVFA: capillary nonperfusion, delayed venous filling, optic disk hyperfluorescence secondary to dilated capillaries, blocked fluorescence secondary to retinal hemorrhages, macular leakage.
OCT: macular edema, optic nerve head evaluation, presence of subretinal fluid.
Pathology
Constriction of the retinal arterioles, arteriosclerosis, hemorrhages, intraretinal lipid, microaneurysms, cytoid bodies.
Disease Course and Natural Progression
There are four grades:
Grade 0 is no changes.
Grade 1 is barely detectable arterial narrowing.
Grade 2 is obvious arterial narrowing with focal irregularities.
Grade 3 is grade 2 plus hemorrhages and/or exudates.
Grade 4 is grade 3 plus disk swelling.
Treatment and Management
Medical therapy to control the elevated blood pressure.
Medications
Systemic antihypertensive medications.
Follow-Up
Variable depending on severity of hypertensive retinopathy.
Differential Diagnosis
BRVO (hemorrhages along a vascular arcade, unilateral, may be bilateral).
CRVO (unilateral flame-shaped, intraretinal hemorrhages in all four quadrants, venous dilation and tortuosity).
Collagen vascular disease (history of polyarteritis nodosa, Wegener granulomatosis, rheumatoid arthritis, systemic lupus erythematosus; abnormal laboratory results may include ANA, anti–double-stranded DNA [anti-dsDNA], anti-ssA, anti-ssB, and c-ANCA).
HIV retinopathy: positive HIV serology.
Leukemia (Roth spots, abnormal peripheral smear, bone marrow biopsy).
Radiation retinopathy (history of radiation therapy).
Sickle cell retinopathy (abnormal sickle cell preparation, hemoglobin electrophoresis, sea-fan NV, peripheral nonperfusion).
Diabetic retinopathy (dot-blot hemorrhages, microaneurysms, NV).
References
Kline R, Kline BE, Moss SE, et al. Blood pressure, hypertension, retinopathy in a population.Trans Am Ophthalmol Soc 1993; 91:207–226.
Tso MOM, Abrams GW, Jampol LM. Hypertensive retinopathy, choroidopathy, and optic neuropathy: clinical and pathophysiological approach to classification. In: Singerman LJ, Jampol LM, eds.Retinal and choroidal manifestations of systemic disease. Baltimore, MD: Williams & Wilkins, 1991.
Embolic Diseases
Summary
Embolic retinal disease is a condition of visible intraretinal deposits from intravascular migration of endogenous cholesterol or calcium or transit of an injected substance.
Etiology
Intra-arterial emboli from any source:
Carotid occlusive disease.
Cardiac valvular disease.
Bone trauma.
Chronic intravenous use of talc, corn starch in drugs.
Steroid embolization after periocular injection or facial injection.
Signs and Symptoms
Asymptomatic or decreased vision.
Demographics
Hypertension.
Carotid occlusive disease.
Cardiac valve disease.
Trauma.
Patients treated for uveitis.
Drug abusers.
Ophthalmic Findings
 Intra-arterial emboli most frequently found at bifurcation of vessels.
Intra-arterial emboli most frequently found at bifurcation of vessels.
 Refractile intravascular particles (Table 3.6).
Refractile intravascular particles (Table 3.6).
Systemic Findings
Carotid bruit.
Cardiac murmur.
Trauma.
Needle tracks.
CVA.
Table 3.6. Features of retinal emboli
Special Tests
Carotid Doppler.
IVFA: refractile intravascular and intraretinal deposits.
OCT: Initially, there is thickening of the inner retina in the region of the of the retina supplied by the obstructed artery. Eventually, the inner retina becomes severely attenuated.
Pathology
Intraretinal deposits of foreign substance.
Disease Course and Natural Progression
Release of endogenous cholesterol, calcium, or lipid; chronic use of talc or corn starch leads to deposition in the retina. Patients are asymptomatic or have decreased vision if there is macular infarction.
Treatment and Management
Send for immediate carotid or cardiac emboli workup.
Cease drug abuse.
Medications
 Cholesterol emboli may be dislodged downstream by ocular massage.
Cholesterol emboli may be dislodged downstream by ocular massage.
 Intraocular pressure (IOP)–lowering agents.
Intraocular pressure (IOP)–lowering agents.
Follow-Up
Per internist to lower cholesterol, control diabetes, lower blood pressure, and stop smoking.
Differential Diagnosis
Platelet fibrin emboli (history of carotid disease, grayish coloration of embolus).
Cholesterol emboli (yellow; vessel bifurcations and carotid disease).
Calcium emboli (white, near disk, cardiac valve disease).
Cardiac myxoma (young patient, left eye more frequent).
Lipid emboli (history of chest trauma, bone fracture, or pancreatitis).
Retrobulbar steroid injection.
Reference
Arruga J, Sanders MD. Ophthalmologic findings in seventy patients with evidence of retinal embolism.Ophthalmology 1982;89:1336–1347.
Hollenhorst Plaques
Summary
Hollenhorst plaques are cholesterol emboli that usually originate from the carotid arteries or major cardiac arteries and lodge in the retinal arterial system.
Etiology
Atherosclerotic deposits in the carotid artery, cardiac arteries, or in the central retinal artery.
Signs and Symptoms
Asymptomatic, transient visual obscurations, unilateral weakness of an arm or leg, transient ischemic attacks, CVA.
Demographics
Embolus is visible in 20% of patients with CRAO.
Ophthalmic Findings
 Glistening yellow cholesterol embolus present at the arterial bifurcation (Figure 3.28).
Glistening yellow cholesterol embolus present at the arterial bifurcation (Figure 3.28).
Systemic Findings
Hypertension.
Diabetes.
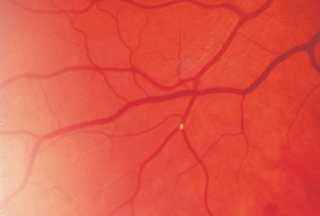
FIGURE 3.28. Hollenhorst plaque at an arteriolar bifurcation.
Special Tests
Color photographs.
Carotid auscultation reveals a bruit.
Carotid Doppler shows narrowing of the carotid artery.
Echocardiogram (transesophageal) may reveal cardiac pathology.
Pathology
Cholesterol embolus in the lumen of the arteriole.
Disease Course and Natural Progression
Fifty-six percent mortality over 9 years versus 27% for age-matched controls without emboli.
Treatment and Management
Carotid endarterectomy.
Control systemic hypertension and diabetes.
Medications
Drugs to lower cholesterol, control diabetes, lower blood pressure, and stop smoking.
Follow-Up
Variable; medical evaluation is most important.
Differential Diagnosis
Calcific emboli: cardiac valve disease, white coloration.
Talc, starch: i.v. drug abuse.
Platelet fibrin emboli: also seen in carotid disease, grayish coloration.
Cardiac myxoma: younger patient, left eye more commonly involved than right eye.
Lipid embolus: history of chest trauma, pancreatitis, or bone fracture.
Reference
Gold DH: Retinal arterial occlusions.Trans Am Acad Ophthalmol Otolaryngol 1977;83:392–408.
Vein Occlusion
Central Retinal Vein Occlusion
Summary
CRVO occurs in an ischemic and nonischemic variety secondary to thrombosis of the central retinal vein at the level of the lamina cribrosa.
Etiology
Thrombosis of the central retinal vein at and posterior to the level of the lamina cribrosa.
Signs and Symptoms
Decreased vision.
May have transient visual obscuration before event.
Demographics
 Ninety percent of patients are over 50 years of age.
Ninety percent of patients are over 50 years of age.
 Association with chronic open-angle glaucoma, hypertension, diabetes, oral contraceptives, and diuretics.
Association with chronic open-angle glaucoma, hypertension, diabetes, oral contraceptives, and diuretics.
Ophthalmic Findings
See Figure 3.29.
Dilated tortuous retinal veins.
Swollen optic disks.
Intraretinal hemorrhages.
Retinal edema.
Cotton-wool spots.
Ischemic CRVO may have a relative afferent pupillary defect.
Systemic Findings
Hypertension is present in 61% of patients.
Diabetes is present in 7%.
Arteriosclerosis.
Special Tests
 IVFA.
IVFA.
Nonischemic CRVO:
Diffuse capillary leakage.
Prolongation of venous filling time.
Ischemic CRVO:
Widespread capillary nonperfusion.
Pronounced prolongation of venous filling time.
 ERG: ischemic type of CRVO; there is decreased ERG bright flash, and a diminished scotopic b:a wave-amplitude ratio (photoreceptors receive nourishment from the unaffected choriocapillaris).
ERG: ischemic type of CRVO; there is decreased ERG bright flash, and a diminished scotopic b:a wave-amplitude ratio (photoreceptors receive nourishment from the unaffected choriocapillaris).
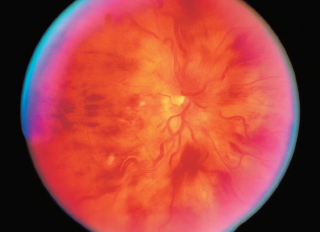
FIGURE 3.29. CRVO with multiple nerve-fiber layer hemorrhages and cotton wool spots in all quadrants, optic disk edema, and dilated tortuous vessels.
 Gonioscopy to evaluate for angle NV.
Gonioscopy to evaluate for angle NV.
Pathology
 Thrombosis of the central retinal vein at the level of the lamina cribrosa.
Thrombosis of the central retinal vein at the level of the lamina cribrosa.
 Acute occlusions present with retinal edema, focal retinal necrosis, and subretinal, intraretinal, and preretinal hemorrhages.
Acute occlusions present with retinal edema, focal retinal necrosis, and subretinal, intraretinal, and preretinal hemorrhages.
 Chronic CRVO is characterized by hemosiderosis, disorganization of the retinal architecture, gliosis, and fibrovascular preretinal membranes.
Chronic CRVO is characterized by hemosiderosis, disorganization of the retinal architecture, gliosis, and fibrovascular preretinal membranes.
Disease Course
 Most cases of nonischemic CRVO (48%) completely resolve.
Most cases of nonischemic CRVO (48%) completely resolve.
 Twenty-two percent progress to complete occlusion.
Twenty-two percent progress to complete occlusion.
 Thirty percent have partial resolution.
Thirty percent have partial resolution.
 Prognosis is markedly worse in eyes with an ischemic CRVO:
Prognosis is markedly worse in eyes with an ischemic CRVO:
Ten percent of patients achieve acuity better than 20/400.
Thirty percent develop neovascularization of the iris (NVI) usually within 3 months (Table 3.7).
Treatment and Management
 Measure IOP.
Measure IOP.
 Perform gonioscopy to see if there is angle closure or angle NV.
Perform gonioscopy to see if there is angle closure or angle NV.
 Perform a dilated fundus examination.
Perform a dilated fundus examination.
 Initially retinal hemorrhage may preclude good IVFA assessment of extent of nonperfusion, but VA is another way to assess this.
Initially retinal hemorrhage may preclude good IVFA assessment of extent of nonperfusion, but VA is another way to assess this.
 Initial poor VA correlates with ischemia.
Initial poor VA correlates with ischemia.
 As the hemorrhage clears, an IVFA may be useful to demonstrate areas of nonperfusion.
As the hemorrhage clears, an IVFA may be useful to demonstrate areas of nonperfusion.
 Consultation with an internist is important to treat associated medical conditions such as hypertension, diabetes, or elevated cholesterol.
Consultation with an internist is important to treat associated medical conditions such as hypertension, diabetes, or elevated cholesterol.
 OCT to assess presence of macular edema
OCT to assess presence of macular edema
Table 3.7. Area of retinal ischemia and risk of developing NVI

NVI, neovascularization of the iris.
 Laser treatment:
Laser treatment:
No benefit of grid photocoagulation for macular edema in older patients with CRVO.
A trend toward improved vision in younger patients treated with focal grid laser in the CRVO study.
PRP if iris NV is present.
Medications
Ozurdex, sustained delivery dexamethasone implant, is the first FDA-approved pharmacologic to treat macular edema secondary to RVO. Other intravitreal anti-VEGF agents and intravitreal steroids have been employed with variable success in treating macular edema secondary to RVO.
Intravitreal anti-VEGF agents can be used as an adjunct to PRP to treat rubeosis.
Follow-Up
Every 6 weeks for the first 6 months; then depending on whether macular edema is being treated.
Differential Diagnosis
Hyperviscosity retinopathy: usually bilateral, history of multiple myeloma or Waldenstrom macroglobulinemia, lymphoma, or leukemia.
Ocular ischemic syndrome: decreased retinal arterial pressure, primarily peripheral intraretinal hemorrhages; carotid occlusion.
Vasculitides: sarcoidosis (elevated ACE, abnormal gallium scan, abnormal CXR, granulomatous uveitis), systemic lupus erythematosus (positive ANA, anti-dsDNA; rheumatologic, dermatologic, pulmonary, or cardiac abnormalities), syphilis (positive result on rapid plasmin reagin [RPR] test and FTA-Abs).
Diabetic retinopathy: hemorrhages, microaneurysms concentrated in posterior pole.
Reference
Central Vein Occlusion Study Group. Baseline and early natural history report: the central vein occlusion study.Arch Ophthalmol 1993;111:1087–1095.
Branch Retinal Vein Occlusion
Summary
BRVOs are occlusions of the vein at the site of AV crossing.
Etiology
Occlusion of the vein at the AV crossing due to atherosclerotic arteriole compressing adjacent vein confined by common adventitial sheath.
Signs and Symptoms
Blurred vision.
May be asymptomatic.
Demographics
Age 60 to 70 years.
Association with hypertension.
Ophthalmic Findings
See Figure 3.30.
 Superficial hemorrhages.
Superficial hemorrhages.
 Retinal edema.
Retinal edema.
 Cotton-wool spots in the involved retinal sector.
Cotton-wool spots in the involved retinal sector.
 Dilated tortuous vein.
Dilated tortuous vein.
 The superotemporal quadrant is involved 63% of the time.
The superotemporal quadrant is involved 63% of the time.
 Sclerosed and sheathed vessels in chronic cases.
Sclerosed and sheathed vessels in chronic cases.
 Collateral vessels that cross horizontal raphe.
Collateral vessels that cross horizontal raphe.
Systemic Findings
Association with systemic hypertension.
Special Tests
IVFA: delayed venous filling in affected vessel, blocked fluorescence due to hemorrhage, nonperfusion, retinal edema, hyperfluorescence secondary to NV.
OCT: to assess presence of macular edema.
Pathology
 Common adventitia binds the branch artery and vein in a normal retina.
Common adventitia binds the branch artery and vein in a normal retina.
 Thickening of the arterial wall (atherosclerosis) compresses the adjacent vein causing turbulence of flow, endothelial damage, and thrombotic occlusion.
Thickening of the arterial wall (atherosclerosis) compresses the adjacent vein causing turbulence of flow, endothelial damage, and thrombotic occlusion.
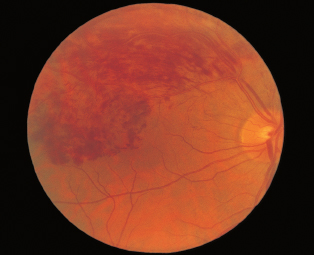
FIGURE 3.30. Superotemporal BRVO.
Disease Course
 Most spontaneously resolve with VA remaining 20/40 or better.
Most spontaneously resolve with VA remaining 20/40 or better.
 Persistent macular edema may lead to decreased vision.
Persistent macular edema may lead to decreased vision.
 Greater than five disk areas of nonperfusion at risk for NV of the retina and VH.
Greater than five disk areas of nonperfusion at risk for NV of the retina and VH.
Treatment and Management
 As the hemorrhage clears, an IVFA may be useful to demonstrate areas of nonperfusion.
As the hemorrhage clears, an IVFA may be useful to demonstrate areas of nonperfusion.
 Focal grid photocoagulation is recommended for chronic macular edema with intact perifoveal capillary perfusion after at least 3 months’ delay for spontaneous resolution of macular edema. Initiate focal grid if VA is 20/40 to 20/200. Retreatment is recommended every 3 months if areas of leakage persist.
Focal grid photocoagulation is recommended for chronic macular edema with intact perifoveal capillary perfusion after at least 3 months’ delay for spontaneous resolution of macular edema. Initiate focal grid if VA is 20/40 to 20/200. Retreatment is recommended every 3 months if areas of leakage persist.
 Intravitreal steroids and intravitreal anti-VEGF agents can be used to treat macular edema.
Intravitreal steroids and intravitreal anti-VEGF agents can be used to treat macular edema.
 PRP if there is retinal or iris NV.
PRP if there is retinal or iris NV.
Medications
Same as CRVO.
Follow-Up
Depends on extent of complications such as macular edema and ischemia.
Differential Diagnosis
Diabetic retinopathy: history of diabetes mellitus, scattered hemorrhages throughout posterior pole, microaneurysms, hard exudates.
Radiation retinopathy: history of local radiation treatment, similar appearance to diabetic retinopathy.
Arteriole macroaneurysm: most common in elderly women, hyperfluorescence of macroaneurysm on IVFA, spontaneous involution of lesion.
References
Branch Vein Occlusion Study Group. Argon laser scatter photocoagulation for prevention of neovascularization and vitreous hemorrhage in branch vein occlusion: a randomized clinical trial.Arch Ophthalmol 1986;104:34–41.
BVOS Group. Argon laser photocoagulation for macular edema in branch vein occlusion.Am J Ophthalmol 1984;98: 271–282.
Central and Branch Retinal Artery Occlusion
Summary
Sudden painless visual loss secondary to occlusion of the central retinal artery or one of its branches.
Etiology
Atherosclerosis-related thrombosis at the level of the lamina cribrosa. Other etiologies include embolization, atherosclerotic plaque, spasm, dissecting aneurysm, hemorrhage under an atherosclerotic plaque, circulatory collapse. One to two percent of patients have giant cell arteritis.
Signs and Symptoms
Profound, sudden, severe painless loss of vision. Some have a history of amaurosis fugax. Sixty-six percent have vision of less than 20/400. Eighteen percent have a vision of ≥20/40 when there is a patent cilioretinal artery. No light perception is evident in patients with combined ophthalmic retinal artery occlusions and CRAOs.
Demographics
Mean age in the early 60s.
No sex predilection.
Right and left eyes affected equally.
Bilateral in 1% to 2% of patients.
Ophthalmic Findings
Afferent pupillary defect.
Opacified and edematous retina. This resolves in 4 to 6 weeks.
Optic disk pallor.
Vascular attenuation, box carring.
Macular cherry-red spot.
Visible embolus in 20% of eyes (Hollenhorst plaque: carotid; calcific plaque: cardiac valve).
Systemic Findings
 Hypertension in two thirds of patients.
Hypertension in two thirds of patients.
 Diabetes in one fourth of patients.
Diabetes in one fourth of patients.
 Cardiac valvular disease in one fourth of patients.
Cardiac valvular disease in one fourth of patients.
 Carotid atherosclerosis in 50% of patients.
Carotid atherosclerosis in 50% of patients.
 The leading cause of subsequent death is cardiovascular disease.
The leading cause of subsequent death is cardiovascular disease.
Special Tests
IVFA: Normal choroidal filling, delayed arterial filling time, and delayed AV transit time. The filling time reverts to normal over time (Figure 3.31).
Erythrocyte sedimentation rate (ESR): Elevated in giant cell arteritis.
ERG: decreased b wave.
Visual field shows a remaining temporal island. If there is a patent cilioretinal artery, the visual field may show a small central island.
Carotid Doppler examinations show carotid stenosis.
Echocardiogram may show valvular plaques.
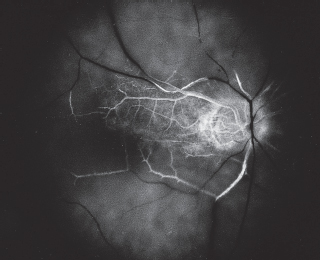
FIGURE 3.31. Late phase of intravenous fluorescein angiogram of a CRAO demonstrating severe retinal nonperfusion.
Pathology
Thrombosis at the level of the lamina scleralis, inner ischemic atrophy.
Disease Course and Natural Progression
 There may be a history of amaurosis fugax.
There may be a history of amaurosis fugax.
 Permanent visual deficit.
Permanent visual deficit.
 Eighteen percent of patients develop NVI.
Eighteen percent of patients develop NVI.
Treatment and Management
From animal studies, efforts to reverse damage following a CRAO are probably ineffective after approximately 90 minutes from the occlusive event.
 Reduce IOP by ocular massage, anterior chamber paracentesis.
Reduce IOP by ocular massage, anterior chamber paracentesis.
 Inhalation therapy with 95% oxygen and 5% carbon dioxide 10 minutes every hour and every 4 hours through the night.
Inhalation therapy with 95% oxygen and 5% carbon dioxide 10 minutes every hour and every 4 hours through the night.
 Oral acetazolamide, aspirin.
Oral acetazolamide, aspirin.
 Intravenous steroids for patients with giant cell arteritis.
Intravenous steroids for patients with giant cell arteritis.
Medications
Intravenous steroids for patients with giant cell arteritis to prevent involvement of fellow eye.
Follow-Up
Every 6 weeks for the first 6 months; as needed thereafter. Administer PRP if NVI develops.
Differential Diagnosis
 Intraocular gentamicin injection.
Intraocular gentamicin injection.
 Arteritic anterior ischemic optic neuropathy: history of jaw claudication, scalp tenderness, weight loss, fever, elevated ESR.
Arteritic anterior ischemic optic neuropathy: history of jaw claudication, scalp tenderness, weight loss, fever, elevated ESR.
 Ophthalmic artery occlusion: no cherry-red spot, entire retina white.
Ophthalmic artery occlusion: no cherry-red spot, entire retina white.
Reference
Hayreh SS, Kolder HE, Weingeist TA. Central retinal artery occlusion and retinal tolerance time.Ophthalmology 1980; 87:75–78.
Ocular Ischemic Syndrome
Summary
Ocular ischemic syndrome is a condition with ocular symptoms and signs attributable to chronic, severe carotid artery or ophthalmic artery obstruction and subsequent ocular hypoperfusion (Table 3.8).
Etiology
Atherosclerosis, giant cell arteritis.
Signs and Symptoms
Gradual visual loss over a period of weeks to months; dull, aching pain; prolonged recovery to bright light.
Demographics
 Age over 55 years with a mean of 65 years of age.
Age over 55 years with a mean of 65 years of age.
 Male more often than female patients by a ratio of 2:1, affects 7.5 per million individuals.
Male more often than female patients by a ratio of 2:1, affects 7.5 per million individuals.
 Greater than 90% obstruction of carotid flow.
Greater than 90% obstruction of carotid flow.
 Twenty percent of patients bilateral involvement.
Twenty percent of patients bilateral involvement.
Ophthalmic Findings
Iris NV with high or low IOP.
Anterior chamber cell and flare.
Narrow retinal arteries.
Dilated veins.
Hemorrhages.
Midperipheral microaneurysms.
NVD.
NVE.
Cotton-wool spots.
Cherry-red spot.
Spontaneous pulsations of the arteries.
Systemic Findings
Hypertension.
Diabetes.
Cardiovascular disease.
CVAs.
Peripheral atherosclerotic vascular disease.
Five-year mortality is 40%.
Table 3.8 Ophthalmikc features that help to distinguish the ocular ischemic syndrome from other ocular diseases.

Special Tests
IVFA: delayed choroidal filling, delayed AV transit time, vascular staining, macular edema (15%), retinal capillary nonperfusion.
Ophthalmodynamometry: decreased retinal artery perfusion pressure.
ERG: decreased amplitude of a and b waves.
Carotid angiography: demonstrates greater than 90% stenosis.
Pathology
Atherosclerotic narrowing of the carotid arteries.
Disease Course and Natural Progression
Progressive carotid stenosis leads to gradual vision loss and dull, aching ocular pain.
Treatment and Management
Carotid endarterectomy: 25% get stabilization or improvement in vision. Surgery is recommended if there is greater than 70% stenosis and the patient is symptomatic. The 2-year CVA rate is 9% for treated versus 26% of untreated patients.
PRP: for NVI.
Medications
None.
Follow-Up
Every 6 weeks and monitor for rubeosis and pressure elevation.
Differential Diagnosis
CRVO: swollen disk, normal retinal artery perfusion pressure, normal choroidal filling, venous staining.
PDR: bilateral, variable age, normal retinal artery perfusion pressure, posterior pole microaneurysms, hard exudate, normal choroidal filling and AV transit, absent vessel staining.
Reference
North American Symptomatic Carotid Endarterectomy Trial Collaborators. Beneficial effect of carotid endarterectomy in symptomatic patients with high grade carotid stenosis.N Engl J Med 1991;325:445–453.
Macroaneurysms
Summary
Acquired macroaneurysms are round or fusiform dilations of the retinal arterioles occurring in the posterior pole within the first three orders of arteriolar bifurcation.
Etiology
Systemic hypertension.
Signs and Symptoms
Asymptomatic or decreased vision.
Demographics
Female more often than male patients.
Ten percent are bilateral, may be multiple.
Age range is 60 to 70 years.
Ophthalmic Findings
See Figure 3.32.
Sub-ILM, intraretinal, subretinal hemorrhages.
Macular edema.
VH.
Arteriolar emboli.
Capillary telangiectasia.
Systemic Findings
Two thirds have hypertension, arteriosclerotic cardiovascular disease.
Special Tests
IVFA: blockage by surrounding hemorrhage. In the early phase, there is arterial filling, and in the late stages, there is staining of the vessel walls due to leakage. There is also leakage of the surrounding capillaries and dilation of the arterial vessel with hyperfluorescence.
OCT: shows location of hemorrhage.
Pathology
Distension of involved retinal arteriole.
Fibroglial proliferation.
Dilated capillaries.
Extravasated blood, lipoidal exudates, hemosiderin deposits.
Disease Course and Natural Progression
 Good visual prognosis, since lesions often thrombose and undergo spontaneous involution with resolution of exudate.
Good visual prognosis, since lesions often thrombose and undergo spontaneous involution with resolution of exudate.
 Some patients have progression of exudation with further vision loss.
Some patients have progression of exudation with further vision loss.
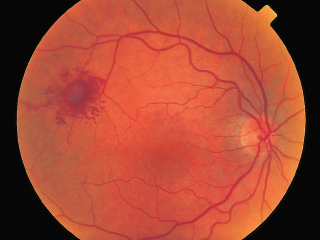
FIGURE 3.32. Macroaneurysm with surrounding retinal hemorrhage.
Treatment and Management
 Laser photocoagulation if macular edema is visually significant. Light treatment with a large spot size is recommended. One should be careful if the distal portion of the arteriole supplies the macula, since a complication of treatment is occlusion of the vessel.
Laser photocoagulation if macular edema is visually significant. Light treatment with a large spot size is recommended. One should be careful if the distal portion of the arteriole supplies the macula, since a complication of treatment is occlusion of the vessel.
 Systemic evaluation for hypertension and cardiovascular disease.
Systemic evaluation for hypertension and cardiovascular disease.
Medications
None.
Follow-Up
Variable depending on macular involvement.
Differential Diagnosis
Diabetic retinopathy: abnormal glucose tolerance test, diffuse hemorrhages, macular exudates, and NV.
Retinal telangiectasia: telangiectatic perifoveal vessels.
Capillary hemangioma: autosomal dominant inheritance, red/orange retinal mass.
Malignant melanoma: B and A scan appearance, double circulation on IVFA.
Hemorrhagic PED in ARMD: drusen, RPE changes, no capillary abnormalities.
Reference
Rabb MF, Gagliano DA, Teskee MP. Retinal arterial macroaneurysms.Surv Ophthalmol 1988;33:73–96.
Perifoveal Telangiectasis
Summary
Perifoveal retinal telangiectasis (PFT) is a developmental retinal vascular disorder characterized by ectasia of the retinal capillaries in which there is irregular capillary dilatation and incompetence. PFT is a condition of microaneurysmal and saccular dilation and capillary nonperfusion of the perifoveal capillaries.
Etiology
Unknown.
Signs and Symptoms
Group 1A: Vision loss is usually in the 20/25 to 20/40 range.
Group 1B: There is minimal vision loss usually in the range of 20/25.
Group 2: There is minimal vision loss with vision usually better than 20/30.
Group 3: Patients may have anywhere from minimal vision loss to legal blindness.
Demographics
PFT is divided into congenital and acquired causes:
Group 1A is unilateral congenital PFT. It occurs in men with a mean age of 40 years.
Group 1B is acquired unilateral idiopathic PFT, which occurs in men with a mean age of 40 years.
Group 2 is acquired bilateral PFT, which affects both men and women with an age range of 50 to 60 years; this is the most common type.
Group 3 is acquired bilateral PFT with capillary nonperfusion, which affects patients mostly in their fifties.
Ophthalmic Findings
 In Group 1A, the temporal half of the macula has telangiectatic vessels with macular exudate and edema.
In Group 1A, the temporal half of the macula has telangiectatic vessels with macular exudate and edema.
 In Group 1B, there are telangiectatic vessels involving the 1 o’clock position at the edge of the FAZ with or without exudate.
In Group 1B, there are telangiectatic vessels involving the 1 o’clock position at the edge of the FAZ with or without exudate.
 Group 2 has bilateral symmetric telangiectatic vessels usually less than 1 disk diameter in extent involving the temporal fovea with minimal edema, mild dilation of the retinal capillaries, and graying of the retina. There are right-angled venules present. There is RPE hyperplasia along the right-angled venules. Some cases have a yellow lesion in the FAZ. CNV is possible.
Group 2 has bilateral symmetric telangiectatic vessels usually less than 1 disk diameter in extent involving the temporal fovea with minimal edema, mild dilation of the retinal capillaries, and graying of the retina. There are right-angled venules present. There is RPE hyperplasia along the right-angled venules. Some cases have a yellow lesion in the FAZ. CNV is possible.
 Group 3 has telangiectatic vessels and an enlarged FAZ with disk pallor present; small gold flecks on retinal surface described by Gass.
Group 3 has telangiectatic vessels and an enlarged FAZ with disk pallor present; small gold flecks on retinal surface described by Gass.
Systemic Findings
In the older patients with bilateral involvement, a glucose tolerance test demonstrates diabetes in one third of patients.
Special Tests
IVFA: Group 1A shows leakage from the telangiectatic vessels. Group 1B has minimal leakage. In Group 2, there is perifoveal capillary leakage, dilated ectatic perifoveal capillaries, and blocked fluorescence in the areas of RPE hyperplasia. In Group 3, there is no leakage from the telangiectatic vessels, and there is an enlarged FAZ.
Pathology
Similar to diabetic microangiopathy with deposits of excess basement membrane in the retinal capillaries.
Disease Course and Natural Progression
Congenital or acquired telangiectatic vessels lead to exudation and edema in Groups 1A and 2 or to nonperfusion in Group 3, which causes vision loss.
Treatment and Management
Group 1A: Photocoagulation may be beneficial. Also, spontaneous resolution is possible.
Groups 1B, 2, and 3: No treatment is recommended. Laser photocoagulation has not been found to improve visual outcome.
Associated CNV is treated with anti-VEGF therapies, similar to other causes of CNV.
Medications
None. Intravitreal anti-VEGF and steroids have been employed with variable success to treat secondary macular edema.
Follow-Up
Variable depending on vision and amount of exudation.
Differential Diagnosis
 BRVO affects capillary bed distal to AV crossing, capillary dilation secondary to collateral pathways, venous outflow.
BRVO affects capillary bed distal to AV crossing, capillary dilation secondary to collateral pathways, venous outflow.
 Radiation retinopathy: multiple abnormal retinal areas, soft exudate, NV, and a history of radiation therapy.
Radiation retinopathy: multiple abnormal retinal areas, soft exudate, NV, and a history of radiation therapy.
 Carotid occlusive disease: iris NV, anterior chamber cell and flare, cherry-red spot, arterial narrowing, venous dilation.
Carotid occlusive disease: iris NV, anterior chamber cell and flare, cherry-red spot, arterial narrowing, venous dilation.
 ARMD: fluorescein angiographic findings, deeper leakage, drusen, RPE changes, no retinal capillary disease.
ARMD: fluorescein angiographic findings, deeper leakage, drusen, RPE changes, no retinal capillary disease.
 Foveal macular dystrophy: yellow lesions resemble PFT Group 2. No telangiectatic vessels or leakage on fluorescein.
Foveal macular dystrophy: yellow lesions resemble PFT Group 2. No telangiectatic vessels or leakage on fluorescein.
 Focal choroiditis may be confused with PFT Group 2 secondary to RPE hyperplasia.
Focal choroiditis may be confused with PFT Group 2 secondary to RPE hyperplasia.
 Diabetic retinopathy: diffuse hemorrhages, cotton-wool spots, microaneurysms, NV.
Diabetic retinopathy: diffuse hemorrhages, cotton-wool spots, microaneurysms, NV.
References
Gass JD, Oyakawa RT. Idiopathic juxtafoveolar retinal telangiectasis.Arch Ophthalmol 1982;100:769–780.
Park DW, Schatz H, McDonald HR, et al. Grid laser photocoagulation for macular edema in bilateral juxtafoveal telangiectasis.Ophthalmology 1997;104:1838–1846.
Familial Exudative Vitreoretinopathy (Fevr)
Summary
FEVR is a bilateral autosomal dominant disorder of peripheral retinal vascular development often associated with retinal traction.
Etiology
It is postulated that there is a genetic defect that induces abnormal development of the hyaloid vascular system.
Signs and Symptoms
Asymptomatic.
May have decreased vision.
May present as an exotropia.
Demographics
Autosomal dominant inheritance.
Bilateral and symmetric.
Ophthalmic Findings
Bilateral involvement.
Avascular zone in the peripheral retina.
TRD.
Retinal folds.
Temporal dragging of the macula.
AV shunts at the margin of vascularized retina, peripheral NV.
VH is rare.
Other findings may include myopia, vitreous “snowflakes,” vitreous bands, and retinal pigmentary changes.
Systemic Findings
None.
Special Tests
 IVFA: avascular border posterior to the ora serrata.
IVFA: avascular border posterior to the ora serrata.
 AV anastomoses that leak fluorescein.
AV anastomoses that leak fluorescein.
Pathology
Vitreous membranes, peripheral vascular occlusion, cholesterol crystals, intraretinal and subretinal hemorrhage, chronic subretinal fluid, contracture and thickening of the retina with dilated telangiectatic vessels.
Disease Course and Natural Progression
The majority of teenage carriers have no visual impairment.
Retinal folds may progress rapidly or slowly, or stabilize.
Rarely vision loss beyond 20 to 30 years of age, usually secondary to rhegmatogenous RD or TRD.
Treatment and Management
Examine family members.
Correct strabismus.
Cryotherapy or laser photocoagulation for NV.
Scleral buckle, pars plana vitrectomy for TRD.
Medications
None.
Follow-Up
Variable.
Differential Diagnosis
ROP: prematurity, oxygen treatment, low birth weight, progressive to the cicatricial stage, or aborts and vascularizes the periphery. The avascular zone is unchanged in FEVR.
Persistent hyperplastic primary vitreous (PHPV): retinal folds in conjunction with remnants of the hyaloid vascular system, retinal dysplasia, elongated ciliary processes, glaucoma, abnormal development in the seventh to the eighth week of gestation versus in the last month of gestation for FEVR.
Reference
Tasmin W, Augsberger JJ, Shields JA, et al. Familial exudative vitreoretinopathy.Trans Am Ophthalmol Soc 1981;79: 211–226.
Retinopathy of Prematurity
Summary
ROP is a proliferative retinopathy of premature and low-birth-weight infants.
Etiology
 In the primary stage, oxygen exposure causes vasoconstriction of incompletely vascularized retina.
In the primary stage, oxygen exposure causes vasoconstriction of incompletely vascularized retina.
 In the secondary stage, there is endothelial proliferation.
In the secondary stage, there is endothelial proliferation.
Signs and Symptoms
Asymptomatic.
Strabismus.
Demographics
 Vision loss in 1,300 children per year in the United States.
Vision loss in 1,300 children per year in the United States.
 Sixty-six percent of infants with birth weight less than 1,250 g affected, and 82% of infants with birth weight of less than 1,000 g.
Sixty-six percent of infants with birth weight less than 1,250 g affected, and 82% of infants with birth weight of less than 1,000 g.
 Prematurity, low birth weight, prolonged oxygen exposure, complicated hospital course, and elevated partial pressure of CO2 (PCO2) are the main risk factors.
Prematurity, low birth weight, prolonged oxygen exposure, complicated hospital course, and elevated partial pressure of CO2 (PCO2) are the main risk factors.
Ophthalmic Findings
Temporal dragging of the macula.
Amblyopia.
Myopia.
Strabismus.
Classification of ROP
 Stage1: Demarcation line.
Stage1: Demarcation line.
 Stage2: Ridge.
Stage2: Ridge.
 Stage3: Ridge and extraretinal fibrovascular proliferation.
Stage3: Ridge and extraretinal fibrovascular proliferation.
 Stage4: Subtoral RD.
Stage4: Subtoral RD.
 Stage5: Total RD.
Stage5: Total RD.
 Plus disease is a disease of stage 3 or worse involving posterior pole.
Plus disease is a disease of stage 3 or worse involving posterior pole.
 Threshold disease is a disease of stage 3 or worse involving five contiguous or eight noncontiguous clock-hour positions of retina in zone 1 or 2 with plus disease.
Threshold disease is a disease of stage 3 or worse involving five contiguous or eight noncontiguous clock-hour positions of retina in zone 1 or 2 with plus disease.
Systemic Findings
Low birth weight.
Cerebral palsy.
Respiratory distress syndrome.
Sepsis.
Necrotizing bowel disease.
Special Tests
IVFA: Peripheral nonperfusion, leakage of dye from neovascular vessels (rarely performed).
FIGURE 3.33. Diagram of the vascular pattern associated with normal immature retina and stage I to stage III ROP.
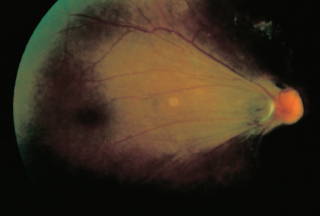
FIGURE 3.34. ROB with temporal dragging of the macula
Pathology
Avascular retina, NV, fibrovascular proliferation in the advanced stages.
Disease Course and Natural Progression
 Eighty-five percent of infants have spontaneous regression.
Eighty-five percent of infants have spontaneous regression.
 Seven percent at weight less than 1,250 g progress to threshold ROP.
Seven percent at weight less than 1,250 g progress to threshold ROP.
 NV leads to fibrosis, which progresses to contracture of proliferative tissue causing RD and macular distortion.
NV leads to fibrosis, which progresses to contracture of proliferative tissue causing RD and macular distortion.
 Examine all high-risk infants, which include those of less than 36 weeks’ gestation or weighing less than 2,000 g.
Examine all high-risk infants, which include those of less than 36 weeks’ gestation or weighing less than 2,000 g.
 Initial examination should be at 7 to 9 weeks of age, and the examination should be performed every 2 weeks until prethreshold value is reached; then examination should be performed every week.
Initial examination should be at 7 to 9 weeks of age, and the examination should be performed every 2 weeks until prethreshold value is reached; then examination should be performed every week.
 Treat when patient reaches threshold, which is defined as stage 3 + ROP in five contiguous or eight cumulative clock-hours of retinal involvement in zone 1 or 2. Treatment is with cryotherapy or laser photocoagulation.
Treat when patient reaches threshold, which is defined as stage 3 + ROP in five contiguous or eight cumulative clock-hours of retinal involvement in zone 1 or 2. Treatment is with cryotherapy or laser photocoagulation.
 Scleral buckle or vitrectomy for stage 4, and pars plana vitrectomy for stage 5.
Scleral buckle or vitrectomy for stage 4, and pars plana vitrectomy for stage 5.
Medications
Anti-vegf therapies (Investigational).
Follow-Up
Every 2 weeks until prethreshold; then every week until regression or threshold is reached, at which time treatment should be started.
Differential Diagnosis
 FEVR: no history of prematurity, family history, no history of oxygen exposure. NV may occur several years after birth.
FEVR: no history of prematurity, family history, no history of oxygen exposure. NV may occur several years after birth.
 Retinoblastoma may resemble stage 4 or 5, calcification on computed tomography (CT).
Retinoblastoma may resemble stage 4 or 5, calcification on computed tomography (CT).
 Congenital cataracts.
Congenital cataracts.
 Norrie disease.
Norrie disease.
Reference
Cryotherapy For Retinopathy Of Prematurity Collaborative Group. Multicenter trial of cryotherapy for retinopathy of prematurity: preliminary results. Arch Ophthalmol 1988;106:471–479.
Coats Disease
Summary
Coats disease is an idiopathic condition characterized by telangiectatic and aneurysmal retinal vessels with intraretinal and subretinal exudates.
Etiology
Primary vascular etiology with breakdown of the blood–retinal barrier versus endocrinologic basis.
Signs and Symptoms
Juvenile type:
Decreased vision.
Strabismus.
Leukocoria.
Adult type:
Asymptomatic.
Decreased vision.
Demographics
Male-to-female ratio of 3:1.
Eighty percent of cases are unilateral.
No racial or ethnic predilection.
Ophthalmic Findings
See Figure 3.35.
Localized yellow/green subretinal exudates.
Serous RD.
Frequently involves macula.
Telangiectatic vessels and microaneurysms.
Systemic Findings
None.
Special Tests
IVFA: numerous localized abnormalities of the retinal vasculature. Early and persistent leakage from telangiectatic vessels and aneurysms. Capillary nonperfusion and anomalous vessels are seen.
OCT: to assess presence of macular edema.
Pathology
 Loss of the vascular endothelium and pericytes with subsequent mural disorganization from ischemia.
Loss of the vascular endothelium and pericytes with subsequent mural disorganization from ischemia.
 Subsequent damage to the blood–retinal barrier results in telangiectasia and massive outpouring of subretinal lipid exudate.
Subsequent damage to the blood–retinal barrier results in telangiectasia and massive outpouring of subretinal lipid exudate.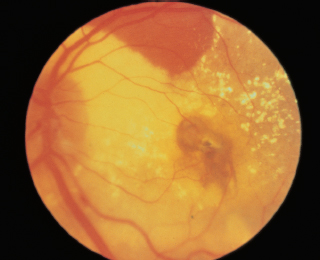
FIGURE 3.35. Coats disease with massive subretinal lipid exudation.
Disease Course and Natural Progression
 Variable but progressive clinical course.
Variable but progressive clinical course.
 Subretinal exudation leads to serous RD.
Subretinal exudation leads to serous RD.
 CNV may develop in the area of the lipid.
CNV may develop in the area of the lipid.
Treatment and Management
 Observation if lesions are limited in extent or do not threaten the macula.
Observation if lesions are limited in extent or do not threaten the macula.
 IVFA-guided photocoagulation if macula is involved or threatened.
IVFA-guided photocoagulation if macula is involved or threatened.
 Cryotherapy if exudative detachment present.
Cryotherapy if exudative detachment present.
 Pars plana vitrectomy if a TRD develops.
Pars plana vitrectomy if a TRD develops.
 Prognosis is poor, and eyes may have amblyopia.
Prognosis is poor, and eyes may have amblyopia.
 Adjunctive intravitreal steroids or intravitreal anti-VEGF agents may be used.
Adjunctive intravitreal steroids or intravitreal anti-VEGF agents may be used.
Medications
None.
Follow-Up
Variable depending on macular involvement.
Differential Diagnosis
Differential diagnosis is that of leukocoria including the following:
Retinoblastoma.
RD.
PHPV.
Congenital cataract.
Norrie disease.
FEVR.
Eales disease.
Vasculitis.
Tumor with exudation.
Diabetes.
BRVO.
Juxtafoveal telangiectasis.
Radiation retinopathy.
Reference
Ridley MD, Shields JA, Brown GC, et al. Coats’ disease: evaluation of management. Ophthalmology 1982;89:1381–1387.
Von Hippel Disease
Summary
Von Hippel disease is an isolated retinal vascular abnormality or one of many systemic abnormalities characterized by a red/orange intraretinal mass with a dilated feeder artery and draining vein.
Etiology
Isolated retinal vascular abnormality.
Signs and Symptoms
Asymptomatic.
Decreased vision.
Demographics
 Autosomal dominant inheritance with incomplete penetrance and sporadic forms occur.
Autosomal dominant inheritance with incomplete penetrance and sporadic forms occur.
 Fifty percent of cases are bilateral.
Fifty percent of cases are bilateral.
Ophthalmic Findings
See Figure 3.36.
 Spherical orange/red tumor fed by a dilated tortuous retinal artery and drained by an engorged vein with associated retinal exudation.
Spherical orange/red tumor fed by a dilated tortuous retinal artery and drained by an engorged vein with associated retinal exudation.
 Lesions are multiple or bilateral in 50% of cases and can involve the retinal or optic nerve.
Lesions are multiple or bilateral in 50% of cases and can involve the retinal or optic nerve.
 Angiomatous variant, which is peripheral and has no large feeding or draining vessel.
Angiomatous variant, which is peripheral and has no large feeding or draining vessel.
Systemic Findings
Von Hippel–Lindau disease (VHL):
Central nervous system (CNS) hemangioblastomas.
Cysts in kidneys, pancreas, liver, epididymis, ovaries.
Renal cell carcinoma.
Pheochromocytoma.
FIGURE 3.36. Von Hippel angioma with dilated feeder vessels.
IVFA: hyperfluorescence of tumor with prominent dilated feeding artery and draining venule (Figure 3.37).
A/B Scan: high internal reflectivity of the angioma.
Pathology
Tortuous large diameter capillaries lined by normal endothelium.
Disease Course and Natural Progression
 Enlargement with time leading to increased exudation, serous detachment, VH, or macular pucker.
Enlargement with time leading to increased exudation, serous detachment, VH, or macular pucker.
 Recurrence after treatment.
Recurrence after treatment.
 May involute spontaneously.
May involute spontaneously.
Treatment and Management
 Photocoagulation or cryotherapy of the lesion. This may result in increased exudation.
Photocoagulation or cryotherapy of the lesion. This may result in increased exudation.
 Pars plana vitrectomy, scleral buckle in cases of RD.
Pars plana vitrectomy, scleral buckle in cases of RD.
 Systemic evaluation for CNS, pancreatic, liver, or renal involvement.
Systemic evaluation for CNS, pancreatic, liver, or renal involvement.
Follow-Up
Variable, depending on extent of exudation.
Differential Diagnosis
Macroaneurysm: hypertension, fusiform dilations of arterioles, with surrounding hemorrhage, exudate.
FIGURE 3.37. IVFA demonstrating hyperfluorescence of the angioma.
Coats disease: telangiectatic aneurysmal vessels, males, unilateral.
Diabetic retinopathy: microaneurysms, dot-blot hemorrhages, concentrated in posterior pole.
Congenital retinal AV malformation.
Sickle cell retinopathy: peripheral nonperfusion, sea-fan NV, sickle preparation.
Presumed acquired retinal hemangioma.
Reference
Hardwig P, Robertson DM. von Hippel-Lindau disease: a familial, often lethal, multisystem phakomatosis. Ophthalmology 1984; 91:263–270.
Wyburn-Mason Disease
Summary
Wyburn-Mason disease is a rare ocular cephalic syndrome in which abnormal retinal AV anastomoses are associated with arteriovenous malformations (AVMs) in the ipsilateral midbrain region.
Etiology
Unknown.
Signs and Symptoms
Mild proptosis.
Poor vision.
Demographics
Sporadic occurrence.
Ophthalmic Findings
See Figure 3.38.
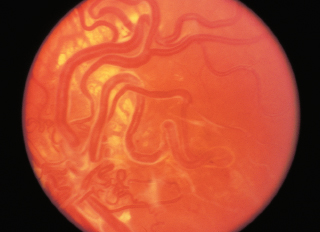
FIGURE 3.38. Racemose angioma with markedly dilated, tortuous anomalous vessels.
 Racemose lesions in the retina, which may also involve the optic nerve, chiasm, tract.
Racemose lesions in the retina, which may also involve the optic nerve, chiasm, tract.
 Usually unilateral.
Usually unilateral.
Systemic Findings
Ipsilateral AVMs of the brain, face, or orbit.
Cranial-nerve palsies.
Visual-field abnormalities.
Special Tests
IVFA: retinal AVM without leakage.
Magnetic resonance imaging (MRI), cerebral angiogram: CNS AVMs commonly involving the midbrain.
Pathology
 Anomalous vessels of the retina with some protruding into the vitreous.
Anomalous vessels of the retina with some protruding into the vitreous.
 Thinning and thickening of vessel walls with fibromuscular medial coats of variable thickness and fibrohyalin adventitial layers.
Thinning and thickening of vessel walls with fibromuscular medial coats of variable thickness and fibrohyalin adventitial layers.
Disease Course and Natural Progression
Usually stationary; however, can rarely cause exudation if the lesion is large.
Treatment and Management
Rarely necessary; cerebral angiography and MRI should be performed to rule out intracranial AVM.
Medications
None.
Follow-Up
Variable.
Differential Diagnosis
VHL: Exudation and exudative RD are more common, autosomal dominant in inheritance.
Reference
Archer DB, Duteman A, Ernest JT, et al. Arteriovenous communications of the retina. Am J Ophthalmol 1973;75:224–241.
Retinal Cavernous Hemangioma
Summary
Cavernous hemangioma is a rare vascular hamartoma composed of clumps of dark intraretinal aneurysms.
Etiology
Unknown.
Signs and Symptoms
Asymptomatic.
Unilateral.
Ten percent are symptomatic when the lesion is located near or in the macula.
Demographics
Questionable autosomal dominant with incomplete penetrance versus variable expression.
Ophthalmic Findings
 Clumps of intraretinal aneurysms, “clusters of grapes.”
Clumps of intraretinal aneurysms, “clusters of grapes.”
 Layering of red blood cells in aneurysms, “pseudohypopyon.”
Layering of red blood cells in aneurysms, “pseudohypopyon.”
 Fibroglial membrane on surface.
Fibroglial membrane on surface.
 VH.
VH.
 Subretinal, intraretinal, preretinal hemorrhages.
Subretinal, intraretinal, preretinal hemorrhages.
Systemic Findings
May have skin or CNS hemangiomas.
Special Tests
IVFA: Aneurysms fill slowly and incompletely; fluorescein collects in the superior portion of the aneurysm with blood blocking fluorescein inferiorly.
Ultrasound: A scan: high internal reflectivity. B scan: irregular surface, large internal acoustic density, absence of choroidal excavation.
Pathology
 Multiple endothelial-lined aneurysms separated by thin fibrous septa.
Multiple endothelial-lined aneurysms separated by thin fibrous septa.
 No intraretinal or subretinal exudate is found.
No intraretinal or subretinal exudate is found.
 Preretinal membrane can overlie the tumor that contains fibrous astrocytes with cytoplasmic filaments.
Preretinal membrane can overlie the tumor that contains fibrous astrocytes with cytoplasmic filaments.
Disease Course and Natural Progression
 Usually stationary; however, visual loss may occur from growth of epiretinal membrane.
Usually stationary; however, visual loss may occur from growth of epiretinal membrane.
 VH is the most common cause of vision loss.
VH is the most common cause of vision loss.
Treatment and Management
Rarely necessary; however, photocoagulation or cryotherapy if VH is present.
Medications
None.
Follow-Up
Variable.
Coats disease: exudative RD.
Leber miliary aneurysms: progressive course.
BRVO: telangiectatic vessels, microaneurysms, exudate.
Capillary hemangioma: feeder vessels, exudate, autosomal dominant inheritance.
Racemose hemangioma: dilation of larger vessels, AV anastomoses.
Diabetic retinopathy: microaneurysms, dot-blot hemorrhages, lipid, exudate, cotton-wool spots, NV.
Reference
Gass JDM. Cavernous hemangioma of the retina: a neuro-oculocutaneous syndrome. Am J Ophthalmol 1971;71: 799–814.
Anemia
Summary
Anemia is a condition of decreased number of normal red blood cells or a reduction of normal hemoglobin that leads to ischemia of the retina and other tissues.
Etiology
Decreased oxygen delivery to the retina.
Signs and Symptoms
Asymptomatic.
Decreased vision.
Floaters.
Demographics
Patients with leukemia, multiple myeloma, Waldenstrom macroglobulinemia, lymphoma, or various chronic diseases can manifest anemia.
Ophthalmic Findings
Superficial flame-shaped hemorrhages.
Cotton-wool spots.
Microaneurysms.
White-centered hemorrhages (Roth spot).
Hard exudates with macular edema
Mild dilation of the arteries and veins.
Arteriolar sheathing in patients with chronic leukemia.
Exudative RD and necrotizing retinitis in patients with Hodgkin disease.
NV.
Systemic Findings
Anemia.
Painless lymph node enlargement.
Reed-Sternberg cells in patients with Hodgkin lymphoma.
Special Tests
IVFA: Capillary nonperfusion, capillary dilation, leakage.
Vitreous cytology for lymphoma cells.
Bone marrow biopsy.
Peripheral smear.
Serum protein electrophoresis (SPEP) to demonstrate plasmacytoma.
Pathology
Hemorrhages, accumulation of axoplasmic debris (cotton-wool spots, capillary occlusion, microaneurysms).
Disease Course and Natural Progression
Ischemia as evidenced by microaneurysms, hard exudate, dilated capillaries, hemorrhages, and arteriolar sheathing may lead to visual loss.
Treatment and Management
Treatment of the underlying condition and anemia.
Medications
Variable depending on condition.
Follow-Up
Medical evaluation is the mainstay of management.
Differential Diagnosis
Hypertensive retinopathy: AV nicking, disk edema.
Diabetic retinopathy: Elevated hemoglobin A1c or abnormal glucose tolerance test. Dot-blot hemorrhages, NV of disk, neovascularization of iris.
Endocarditis: blood cultures, echocardiogram.
Collagen vascular disease: Polyarteritis Nodosum, Wegener Granulomatosis, Systemic Lupus Erythematosus, Rheumatoid Arthritis: history of arthritis, skin rashes or evidence of systemic vasculitis, abnormal laboratory test results: ANA, c-ANCA, anti-ssA, anti-ssB, anti-dsDNA, rheumatoid factor.
Radiation retinopathy: history of radiation treatment, microaneurysms.
Sickle cell retinopathy: peripheral nonperfusion, sea-fan NV.
Reference
Kincaid MC, Green WR. Ocular and orbital involvement in leukemia. Surv Ophthalmol 1983;27:211–232.
Hyperviscocity
Summary
Increased blood viscosity causes accumulation of blood products and cells leading to vascular occlusion.
Leukemia.
Lymphoma.
Waldenstrom macroglobulinemia.
Polycythemia vera.
Multiple myeloma.
Signs and Symptoms
Asymptomatic; patients with lymphoma may have bleeding diatheses and organomegaly.
Demographics
 Seventy-five percent of chronic leukemias have intraocular involvement.
Seventy-five percent of chronic leukemias have intraocular involvement.
 Eighty-two percent of acute leukemias have intraocular involvement.
Eighty-two percent of acute leukemias have intraocular involvement.
Ophthalmic Findings
White-centered hemorrhages.
Choroidal infiltrates.
Serous RDs.
Hemorrhages in all retinal layers.
Cotton-wool spots.
Venous occlusion.
Microaneurysms.
NV (chronic myelogenous leukemias).
Venous dilation.
Papilledema (polycythemia vera).
Pars plana cysts (multiple myeloma).
Sea-fan NV may be present.
Systemic Findings
 CNS leukemia.
CNS leukemia.
 Acute leukemia patients may have bleeding diatheses and infections.
Acute leukemia patients may have bleeding diatheses and infections.
 Vague symptoms in patients with chronic leukemias.
Vague symptoms in patients with chronic leukemias.
 Increased immunoglobulin M (IgM) fraction of plasma proteins in patients with Waldenstrom macroglobulinemia.
Increased immunoglobulin M (IgM) fraction of plasma proteins in patients with Waldenstrom macroglobulinemia.
Special Tests
Bone marrow biopsy.
Peripheral blood smear.
Cytologic examination of the vitreous (reticulum cell carcinoma).
SPEP.
Pathology
 Hemorrhages, neurofibrillar infarcts, venous occlusions, leukemic cells.
Hemorrhages, neurofibrillar infarcts, venous occlusions, leukemic cells.
 Neoplastic infiltrates of the retina and optic nerve in lymphoma.
Neoplastic infiltrates of the retina and optic nerve in lymphoma.
 Low platelet counts are associated with intraretinal hemorrhages.
Low platelet counts are associated with intraretinal hemorrhages.
Disease Course and Natural Progression
Hyperviscosity leads to venous occlusion, microaneurysms, hemorrhages, and peripheral nonperfusion, which stimulates the development of NV.
Treatment and Management
 Chemotherapy, radiation therapy for leukemia.
Chemotherapy, radiation therapy for leukemia.
 Leukopheresis for hyperleukocytic retinopathy.
Leukopheresis for hyperleukocytic retinopathy.
 Plasmapheresis for Waldenstrom macroglobulinemia.
Plasmapheresis for Waldenstrom macroglobulinemia.
Medications
Medical management by a hematologist/oncologist is required.
Follow-Up
Medical evaluation and treatment are most important.
Differential Diagnosis
CRVO: unilateral flame-shaped hemorrhages, venous dilation, tortuosity.
Sickle cell retinopathy: abnormal sickle preparation, hemoglobin electrophoresis, sea-fan NV, peripheral nonperfusion.
Diabetic retinopathy: dot-blot hemorrhages, NV, abnormal glucose tolerance test result, elevated hemoglobin A1c.
Hypertensive retinopathy: AV nicking, elevated blood pressure.
Collagen vascular disease: Polyarteritis Nodosum, Wegener Granulomatosis, Systemic Lupus Erythematosus, Rheumatoid Arthritis: history of arthritis, skin rashes or evidence of systemic vasculitis, abnormal laboratory test results: ANA, c-ANCA, anti-ssA, anti-ssB, anti-dsDNA, rheumatoid factor.
Reference
Carr RE, Henkind P. Retinal findings associated with serum hyperviscosity. Am J Ophthalmol 1963;56:23–31.
Toxoplasmosis
Summary
Worldwide cat-transmitted parasitic zoonosis ranging from life-threatening congenital infection to flulike acquired infection; retinochoroiditis is generally self-limited but may be recurrent, and vision loss is dependent on extent and area of involvement.
 Toxoplasma gondii, an obligate intracellular parasite, exists as trophozoite, bradyzoite, or sporozoite; sporozoites (oocysts) are produced within the cat host by both asexual and sexual reproduction and shed by the millions in feces.
Toxoplasma gondii, an obligate intracellular parasite, exists as trophozoite, bradyzoite, or sporozoite; sporozoites (oocysts) are produced within the cat host by both asexual and sexual reproduction and shed by the millions in feces.
 Oocysts in contaminated meat, poultry, eggs, or dirt are ingested by humans, leading to proliferation of the invasive tachyzoite and its destructive inflammatory sequelae.
Oocysts in contaminated meat, poultry, eggs, or dirt are ingested by humans, leading to proliferation of the invasive tachyzoite and its destructive inflammatory sequelae.
 Tachyzoite invasion of the retina leads to retinitis until the host immune response quells the infection and the parasite is encysted.
Tachyzoite invasion of the retina leads to retinitis until the host immune response quells the infection and the parasite is encysted.
Signs and Symptoms
Unilateral floaters.
Metamorphopsia.
Decreased vision.
Ocular pain and redness.
Flulike symptoms in acquired infection.
Congenital infection: triad of convulsions, cerebral calcifications, and retinochoroiditis; also can have hydrocephalus, mental retardation, jaundice, rash, and fever.
Demographics
 Twenty to seventy percent of persons in the United States have positive titers for toxoplasma. Antibody positivity increases with age.
Twenty to seventy percent of persons in the United States have positive titers for toxoplasma. Antibody positivity increases with age.
 Unclear if majority of cases represent reactivation of congenital infection or if they are acquired.
Unclear if majority of cases represent reactivation of congenital infection or if they are acquired.
 Maternal infection before or during gestation leads to fetal transmission, with severe consequences in first trimester but highest transmission (40%) within the third trimester; bilateral congenital retinochoroiditis occurs in nearly 80% of patients.
Maternal infection before or during gestation leads to fetal transmission, with severe consequences in first trimester but highest transmission (40%) within the third trimester; bilateral congenital retinochoroiditis occurs in nearly 80% of patients.
 More common and virulent in immunocompromised individuals.
More common and virulent in immunocompromised individuals.
Ophthalmic Findings
Fine stellate endothelial keratic precipitates.
Granulomatous anterior chamber inflammatory reaction.
Cataract.
Moderate-to-severe vitritis, “headlight in fog”; precipitates on posterior hyaloid face (Figure 3.39).
Single or multiple patches of necrotizing retinitis, often adjacent to preexisting chorioretinal scar.
Optic neuritis.
CME.
Retinal vasculitis.
Papillitis.
BRVO in areas of retinitis.
Scleritis (rarely).
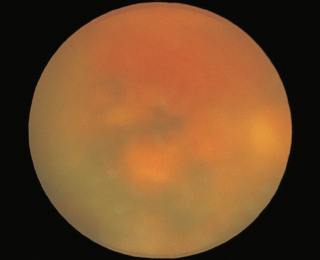
FIGURE 3.39. Acute toxoplasma retinochoroiditis with dense vitritis.
Systemic Findings
Acquired infection: See above.
Congenital infection: See above.
Special Tests
Toxoplasma immunoglobulin G (IgG) and IgM titers.
Sabin-Feldman dye test: tests ability of serum antibody to fix complement and lyse cells.
Enzyme-linked immunosorbent assay (ELISA) or indirect fluorescent antibody test.
Polymerase chain reaction (PCR) of vitreous specimen.
Other tests: FTA-Abs, purified protein derivative (PPD), ELISA for Toxocara organisms.
Pathology
 Retinal necrosis with mononuclear cellular infiltrate; adjacent choroidal necrosis may be seen.
Retinal necrosis with mononuclear cellular infiltrate; adjacent choroidal necrosis may be seen.
 Scars bordering areas of active infection common; cysts or tachyzoites may be identified in or near affected retina.
Scars bordering areas of active infection common; cysts or tachyzoites may be identified in or near affected retina.
Disease Course
 Reactivated retinitis has course of weeks to months, resolving spontaneously.
Reactivated retinitis has course of weeks to months, resolving spontaneously.
 Before resolution, necrotizing macular retinitis may cause permanent visual loss.
Before resolution, necrotizing macular retinitis may cause permanent visual loss.
 Multidrug treatment of active retinitis or papillitis limits spread of infection and may prevent severe visual loss.
Multidrug treatment of active retinitis or papillitis limits spread of infection and may prevent severe visual loss.
 Approximately 80% of patients will have recurrence within 5 years.
Approximately 80% of patients will have recurrence within 5 years.
Treatment and Management
Medical treatment of macular-threatening lesions or cases with significant visual loss due to vitritis.
Triple therapy:
Sulfadiazine: 2 g by mouth (p.o.) × 1, then 1 g q.i.d. with high fluid intake.
Pyrimethamine (Daraprim): 50 mg p.o. Q12 × 2, then 25 mg b.i.d.
Folinic acid (leucovorin): 3 to 5 mg p.o. twice weekly.
Clindamycin, azithromycin, clarithromycin, spiramycin, minocycline, atovaquone, and trimethoprim/sulfamethoxazole are alternative/adjuvant therapies.
Prednisone: 20 to 80 mg p.o. daily can be added to reduce inflammation, only after beginning antibiotics
Infected pregnant women: sulfadiazine, clindamycin, or spiramycin.
Follow-Up
 Weekly; observe for complications of medical treatment (pseudomembranous colitis, bone marrow suppression, steroid effects).
Weekly; observe for complications of medical treatment (pseudomembranous colitis, bone marrow suppression, steroid effects).
 Biweekly complete blood cell count (CBC).
Biweekly complete blood cell count (CBC).
Differential Diagnosis
Syphilis.
Sarcoidosis.
Tuberculous chorioretinitis.
CMV retinitis.
Acute retinal necrosis/Progressive outer retinal necrosis (PORN).
Ocular toxocara.
Intraocular Lymphoma.
Optic neuritis.
Retinoblastoma.
Reference
Engstrom RE Jr, Holland GN, Nussenblatt RB, et al. Current practices in the management of ocular toxoplasmosis. Am J Ophthalmol 1991;111:601–610.
Sarcoidosis
Summary
Sarcoidosis is a multisystem, granulomatous disorder of unknown etiology with ocular involvement occurring in 15% to 25% of patients.
Etiology
Unknown.
Signs and Symptoms
Pain.
Photophobia.
Blurred vision.
Floaters.
Red Eyes.
Demographics
 Most often affects adults younger than 40 years of age.
Most often affects adults younger than 40 years of age.
 Increased incidence in African Americans and native Scandinavians.
Increased incidence in African Americans and native Scandinavians.
 Slight female predilection.
Slight female predilection.
Ophthalmic Findings
 Chronic granulomatous or nongranulomatous anterior uveitis (two third of patients).
Chronic granulomatous or nongranulomatous anterior uveitis (two third of patients).
 Mutton fat keratic precipitates.
Mutton fat keratic precipitates.
 Koeppe and Busacca iris nodules.
Koeppe and Busacca iris nodules.
Posterior involvement in 20% to 30% of patients includes the following:
 Vitritis with white cell clumping (“snowballs”).
Vitritis with white cell clumping (“snowballs”).
 Exudates and sheathing along peripheral retinal vasculature (“candle-wax drippings”).
Exudates and sheathing along peripheral retinal vasculature (“candle-wax drippings”).
 Chorioretinal granulomas.
Chorioretinal granulomas.
 CME.
CME.
 Optic nerve granulomas, disk edema.
Optic nerve granulomas, disk edema.
 Occlusive retinal vasculitis with retinal NV.
Occlusive retinal vasculitis with retinal NV.
Other ocular findings:
 Lacrimal infiltration with keratitis sicca.
Lacrimal infiltration with keratitis sicca.
 Extraocular muscle infiltration with motility restriction and proptosis.
Extraocular muscle infiltration with motility restriction and proptosis.
 Conjunctival granulomas.
Conjunctival granulomas.
 Orbital inflammation.
Orbital inflammation.
Systemic Findings
 May affect lungs, musculoskeletal system, nervous system, skin, spleen, bones, CNS.
May affect lungs, musculoskeletal system, nervous system, skin, spleen, bones, CNS.
 Ninety percent with intrathoracic involvement ranging from hilar lymphadenopathy to diffuse pulmonary infiltration with fibrosis.
Ninety percent with intrathoracic involvement ranging from hilar lymphadenopathy to diffuse pulmonary infiltration with fibrosis.
Special Tests
 Abnormal CXR with evidence of hilar lymphadenopathy.
Abnormal CXR with evidence of hilar lymphadenopathy.
 Biopsy of skin, palpable lymph nodes, lung, liver, and conjunctiva showing noncaseating granulomas is usually required for diagnosis.
Biopsy of skin, palpable lymph nodes, lung, liver, and conjunctiva showing noncaseating granulomas is usually required for diagnosis.
 ACE detects total body granuloma content but is nonspecific.
ACE detects total body granuloma content but is nonspecific.
 Gallium scan of head, neck, and mediastinum for foci of uptake.
Gallium scan of head, neck, and mediastinum for foci of uptake.
 Serum lysozyme, SPEP, and serum calcium.
Serum lysozyme, SPEP, and serum calcium.
 Kveim skin test (nodule induction using a suspension of human sarcoid tissue) is no longer used.
Kveim skin test (nodule induction using a suspension of human sarcoid tissue) is no longer used.
Characteristic noncaseating granuloma composed of epithelioid cells and multinucleated giant cells, often surrounded by a thin, incomplete rim of lymphocytes.
Disease Course
 Three characteristic courses (monophasic, relapsing, or chronic) associated with progressively worsening visual prognosis.
Three characteristic courses (monophasic, relapsing, or chronic) associated with progressively worsening visual prognosis.
 Visual loss often associated with vascular occlusion, retinal NV, macular involvement, or optic nerve lesions.
Visual loss often associated with vascular occlusion, retinal NV, macular involvement, or optic nerve lesions.
 Secondary glaucoma is a poor prognostic sign.
Secondary glaucoma is a poor prognostic sign.
Treatment and Management
 Steroids (topical, depot, systemic) depending on degree of inflammation and response to previous treatment.
Steroids (topical, depot, systemic) depending on degree of inflammation and response to previous treatment.
 Cycloplegics for comfort and prevention of synechiae.
Cycloplegics for comfort and prevention of synechiae.
 Treatment for glaucoma depends on etiology (inflammatory, steroid induced, angle closure, or neovascular).
Treatment for glaucoma depends on etiology (inflammatory, steroid induced, angle closure, or neovascular).
 Steroid-sparing agents required in those with chronic progressive disease especially with posterior involvement.
Steroid-sparing agents required in those with chronic progressive disease especially with posterior involvement.
Medications
Topical steroids.
Periocular steroids.
Systemic steroids for posterior involvement or severe anterior uveitis.
Cycloplegic agents.
Steroid-sparing agents including methotrexate, cyclosporine, azathioprine, mycophenoloate mofetil in those with chronic disease.
Follow-Up
 One to two times per week during acute episodes with cycloplegic and steroid taper as inflammation resolves.
One to two times per week during acute episodes with cycloplegic and steroid taper as inflammation resolves.
 Closely follow IOP and fundus findings.
Closely follow IOP and fundus findings.
 Asymptomatic adults should be seen every 6 months.
Asymptomatic adults should be seen every 6 months.
 Asymptomatic children should be seen every 3 months.
Asymptomatic children should be seen every 3 months.
Differential Diagnosis (Entities and Distinguishing Features)
Tuberculosis: positive PPD, abnormal CXR, acid-fast bacilli on sputum sample or bronchial washing.
Syphilis: positive FTA-Abs or RPR test result.
Idiopathic pars planitis: vitreous opacification with exudate at ora serrata; less significant anterior segment findings. Negative systemic workup and negative review of systems.
Vogt-Koyangi-Harada disease—poliosis, vitiligo, headaches, exudative RD, pigmentary changes, negative CXR, negative laboratory findings.
Reference
Karma A, Huhti E, Poukkula A. Course and outcome of ocular sarcoidosis. Am J Ophthalmol 1988;106:467–472.
Birdshot Retinochoroidopathy
Summary
Birdshot retinochoroidopathy is an ocular inflammatory disease characterized by multiple, bilateral, often symmetric foci of hypopigmentation at the level of the choroid.
Etiology
Possible genetic predisposition in HLA-A29 patients.
Signs and Symptoms
Blurred vision.
Floaters.
Nyctalopia.
Dyschromotopsia.
Demographics
Occurs in third to sixth decade, average age of 50.
Men and women equally affected.
Strong association with HLA-A29 antigen.
Ophthalmic Findings
See Figure 3.40.
 Discrete, creamy, oval, hypopigmented spots less than 0.5 mm in diameter at the level of choroid that are located predominantly posteriorly and nasally.
Discrete, creamy, oval, hypopigmented spots less than 0.5 mm in diameter at the level of choroid that are located predominantly posteriorly and nasally.
 Vitreous inflammation varies from mild to severe. Mild anterior segment inflammation.
Vitreous inflammation varies from mild to severe. Mild anterior segment inflammation.
 May have attenuation of retinal vasculature with inflammatory sheathing.
May have attenuation of retinal vasculature with inflammatory sheathing.
 CME.
CME.
 Epiretinal membrane.
Epiretinal membrane.
 CNVM.
CNVM.
 Disk edema or atrophy may be present.
Disk edema or atrophy may be present.
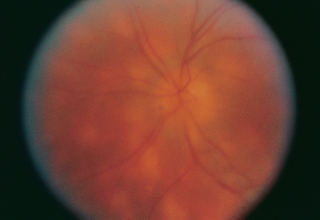
FIGURE 3.40. Birdshot retinochoroidopathy with multiple small yellow choroidal lesions and a mild vitritis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


