Childhood Glaucomas: Classification and Examination
Childhood glaucomas constitute a rare, heterogeneous group of diseases. Often vision-threatening, these diseases present special challenges in diagnosis and optimal management. Parents and primary care providers usually first recognize the abnormalities that lead to the diagnosis of glaucoma in infants and young children, with devastating consequences when that correct diagnosis is substantially delayed. The clinical presentation of glaucoma varies with the age of onset and the severity of intraocular pressure (IOP) elevation. In addition, detailed ophthalmic examination of young children can be difficult, and management strategies for this rare condition are less familiar than those in adult patients. Pharmacologic, technological, and genetic advances in the diagnosis and treatment of glaucoma engender hope that children with this disease may face brighter visual futures.
CLASSIFICATION OF CHILDHOOD GLAUCOMAS
The glaucomas of childhood have been categorized in various ways, but one simple, commonly used system considers them as either primary or secondary in mechanism. Although this classification system is far from ideal (see Chapter 7), we use this terminology here because our somewhat limited insights today preclude a more meaningful conceptual framework for discussing childhood glaucomas.
The primary glaucomas, often genetic in origin, comprise those in which a developmental abnormality of the anterior chamber angle leads to obstruction of aqueous outflow. Within these primary glaucomas, congenital open-angle glaucoma—often termed primary congenital glaucoma (PCG)—and juvenile open-angle glaucoma present without consistently associated ocular or systemic developmental anomalies. By contrast, primary glaucomas associated with ocular abnormalities—often called developmental glaucomas—include primary glaucomas in which a developmental abnormality is responsible for the glaucoma, but in which additional ocular and systemic anomalies are typically present. Unlike the primary childhood glaucomas, secondary childhood glaucomas include glaucomas whose mechanism of outflow obstruction is acquired from other events, such as inflammation or neoplasia, rather than a primary anomaly of the angle. In a yearlong prospective study of all new childhood glaucoma cases in the United Kingdom and the Republic of Ireland, 99 cases were identified: 47 primary and 52 secondary in nature (1).
In this chapter, we consider the pediatric glaucoma patient and details of the examination specific to the child with known or suspected glaucoma. The following chapter, Chapter 14, addresses primary glaucomas of childhood, including PCG and the major developmental glaucomas. Because children are subject to many of the same secondary glaucomas as adults, these topics are discussed together in subsequent chapters of Section II, with special attention given to situations that apply only to children. One exception is the glaucoma occurring after removal of cataracts in infants and young children, as this “aphakic glaucoma” or “pseudophakic glaucoma” may well represent the second most commonly encountered single type of childhood glaucoma (after PCG) (1), and is therefore included in Chapter 14.
Table 13.1 (2) gives one scheme for considering childhood glaucomas (2). Some pediatric glaucomas may have both primary and secondary causes (e.g., infantile-onset glaucoma in Sturge–Weber syndrome, neurofibromatosis, and aniridia). Many of the primary and developmental glaucomas are genetic in origin (see Chapters 8 and 14). Continuing elucidation of the genetics behind many conditions associated with pediatric glaucoma will no doubt lead to replacement of the current phenotypically driven diagnostic labels, with names and categories based on underlying genetic abnormalities.


SIGNS AND SYMPTOMS OF GLAUCOMA IN CHILDHOOD
The signs and symptoms of glaucoma in children vary tremendously with the age of onset and the degree of IOP elevation. Infants and young children with glaucoma (usually with PCG, but occurring with early-onset glaucoma of any cause) usually present because the family or pediatrician has noticed something abnormal about the eyes or the infant’s behavior. Corneal enlargement or opacification (resulting from stretching due to high IOP), or both, often signal glaucoma in the infant; both may progress rapidly over the first 2 years of life if IOP remains elevated (Figs. 13.1 and 13.2). Buphthalmos is a term to describe the abnormal enlargement of an infant’s eye secondary to elevated IOP; in extreme cases, these eyes are vulnerable to lens subluxation and rupture with even minor trauma (Fig. 13.3). The classic triad of findings usually ascribed to PCG (see also Chapter 14)—epiphora, photophobia (Fig. 13.4), and blepharospasm (3)—results from corneal edema, often with associated breaks in the Descemet membrane called Haab striae. Descemet membrane breaks appear to occur only in the first 2 years of life; they leave permanent evidence of early-onset glaucoma and vary with respect to the associated corneal distortion and scarring (Figs. 13.5 and 13.6). Breaks with more vertical orientation may be seen after forceps delivery (Fig. 13.7) (4).
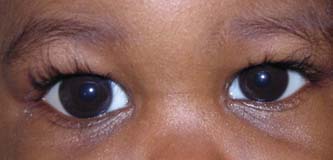
Figure 13.1 Infant with congenital glaucoma, showing buphthalmos and asymmetric enlargement of the corneas, right more than left. Corneal edema has resolved after successful angle surgery.
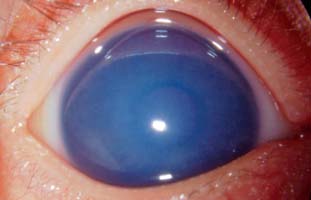
Figure 13.2 Right eye of a 3-month-old infant presenting with enlarged, cloudy cornea in the setting of newly diagnosed congenital glaucoma. IOP was 35 mm Hg.
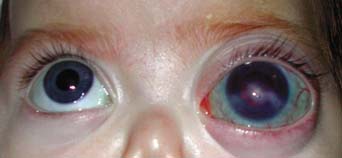
Figure 13.3 Severe buphthalmos in the left eye of a 6-monthold infant with congenital glaucoma and bilateral enlarged corneas and high myopia in the setting of Stickler–Marshall syndrome. This blind left eye was exposed and presumed painful and was subsequently enucleated.

Figure 13.4 Extreme photophobia in an infant girl, 6 months of age, with PCG. Bilateral corneal edema and photophobia improved after surgical treatment.

Figure 13.5 Slitlamp appearance of tears in Descemet membrane, or Haab striae, in a patient with congenital glaucoma.
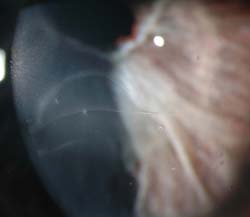
Figure 13.6 Haab striae in a patient with Axenfeld–Rieger glaucoma, viewed at the slitlamp under high magnification.
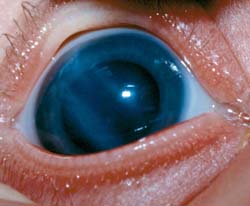
Figure 13.7 Forceps-related tears in Descemet membrane. Note the very straight Haab striae, oriented from superotemporal to inferonasal in the cornea of the right eye of this newborn infant boy. Permanent scarring and high astigmatism resulted in amblyopia in this eye.
Additional nonspecific signs of glaucoma in early life include a deep anterior chamber and optic nerve cupping. In the absence of optic atrophy, the optic cup may decrease greatly in size with IOP reduction and will enlarge again if control of IOP is lost (3). Optic atrophy, which may result from chronic or severe IOP elevation, is irreversible.
In contrast to infants and very young children, older children with glaucoma typically present with decreased vision (usually from induced myopia, but occasionally from end-stage optic nerve damage) or because they are known glaucoma suspects (e.g., with Sturge–Weber syndrome, aniridia, or aphakia or pseudophakia). Although elevated IOP produces corneal enlargement limited to the first 3 years of life, scleral stretching persists for approximately 10 years, producing progressive myopia (and often astigmatism), usually seen in older children with glaucoma. While optic nerve cupping is not by itself a reliable indicator of glaucoma, its presence should prompt a thorough evaluation for possible glaucoma in a child of any age (Fig. 13.8). Older children infrequently present with symptoms of acute glaucoma, such as nausea associated with eye pain, headaches, and even colored haloes around lights (e.g., secondary to trauma or angle closure, as with cicatricial retinopathy of prematurity).
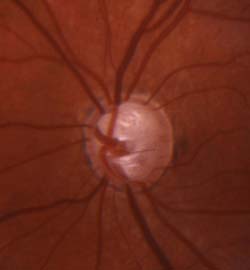
Figure 13.8 Severe optic nerve cupping in the left eye of an 8-year-old girl with juvenile-onset glaucoma.
Visual loss from infant and childhood glaucoma most often results from pathologic changes in the eye, such as corneal opacification and optic nerve damage. Poor vision may also occur despite adequate IOP control, secondary to the development of anisometropia or strabismic amblyopia, especially in unilateral or asymmetric bilateral childhood glaucoma.
DIFFERENTIAL DIAGNOSIS
The clinical features of glaucoma in infancy and childhood overlap partly with those of other pediatric ophthalmic conditions, with the exception of elevated IOP (Table 13.2) (3,5). When faced with ocular signs or symptoms suggestive of possible glaucoma, the clinician must consider and rigorously exclude glaucoma, keeping in mind that identifying a coexisting nonglaucomatous disorder does not eliminate the possibility of glaucoma. For example, glaucoma may complicate uveitis, Peters anomaly, and megalocornea; glaucoma may even occur coincident with the commonly encountered congenital nasolacrimal duct obstruction (3).

THE DIAGNOSTIC EXAMINATION
Although any child with suspected glaucoma requires a detailed pediatric ophthalmic examination, there are specific goals of the glaucoma-related examination: (a) confirming or excluding the diagnosis of glaucoma, (b) determining the cause of the glaucoma (if present), and (c) gathering information (including any prior glaucoma interventions) vital to plan the optimal management. Examination under anesthesia may be avoided if the diagnosis of glaucoma can be confidently excluded (in an infant or young child) or if an older child would benefit from a medication trial. The examination under anesthesia, when it is indicated, provides a one-stop opportunity for more detailed gonioscopy and evaluation of the optic nerve head, as well as measurements of corneal diameter, central corneal thickness, and axial length, immediately followed by any needed surgical intervention.
Vision Testing (Acuity and Visual Fields)
Optimal vision-testing methods will vary with the patient’s age and cognitive function. While central, maintained fixation behavior and absent nystagmus are encouraging in infants, older children should perform optotype testing with proper refractive correction. Visual loss in children with glaucoma often results from ocular changes related to glaucoma or from amblyopia in asymmetric cases; visual acuity loss resulting from optic nerve damage represents an unfortunate, often end-stage situation.
Visual field testing, especially quantitative automated static perimetry, often proves challenging for young children and for all children with nystagmus or poor vision. Hence, perimetry rarely makes the diagnosis of glaucoma but serves instead to assess adequacy of control in older children with glaucoma who can perform reliable baseline examination. Visual field assessment is nonetheless worthwhile in all children with glaucoma, because even confrontation visual fields can often verify suspected severe nasal field loss in children with severe glaucoma and poor vision. Children with associated neurologic conditions (e.g., Sturge–Weber syndrome) may have underlying homonymous hemifield loss independent of their glaucoma. Newer, faster testing algorithms may allow younger children to undergo automated (Humphrey) visual field testing more reliably (6) (Fig. 13.9). Frequency-doubling perimetry (see Chapter 5) may also hold promise for screening and following visual fields over time in children with known or suspected glaucoma (7,8).
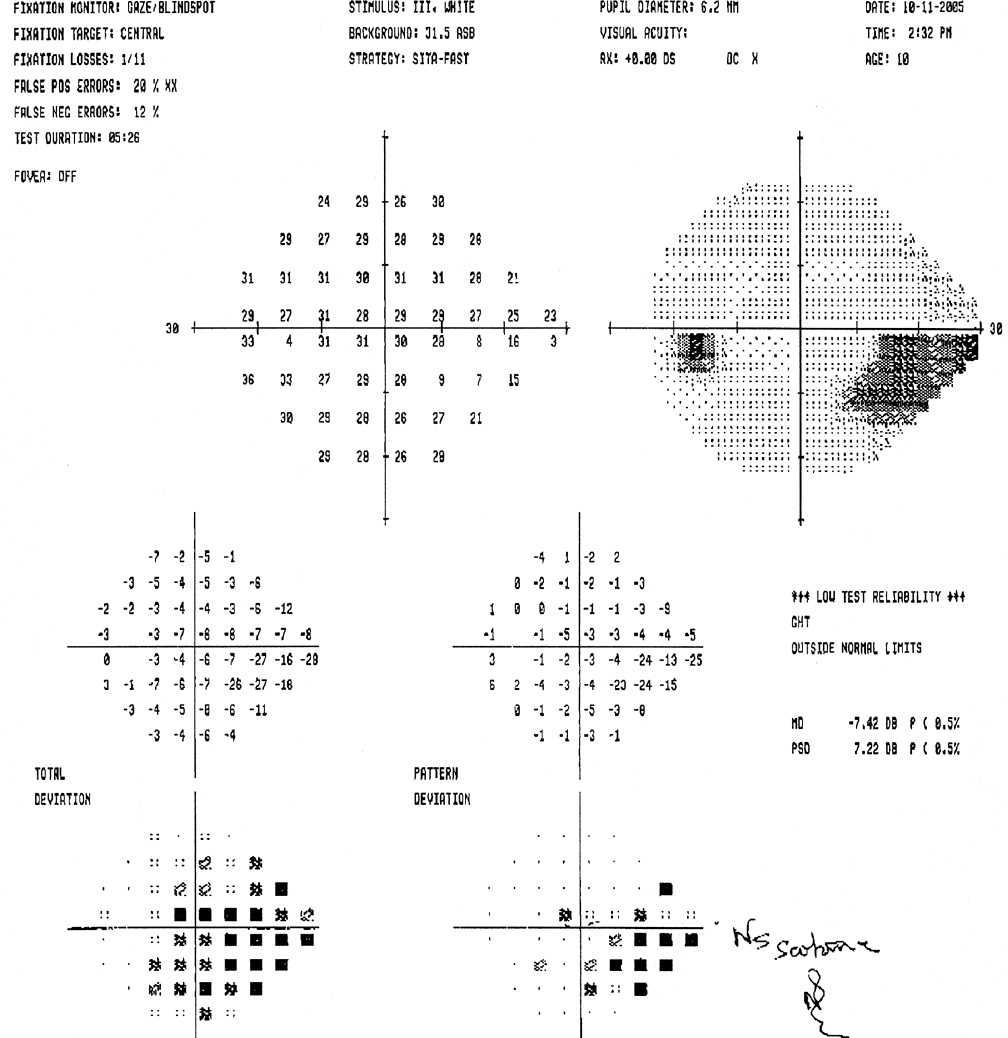
Figure 13.9 Humphrey visual field testing demonstrates an inferior arcuate scotoma in the left eye of this 11-year-old with juvenile open-angle glaucoma, who presented with severe optic nerve cupping and had successful control of glaucoma with mitomycin C–augmented trabeculectomy.
External Examination
External examination helps identify evidence of associated abnormalities (e.g., neurofibromatosis, facial hemangioma), buphthalmos (especially asymmetry between the eyes), photophobia, or nasolacrimal obstruction. Overall assessment of the child’s health and systemic features can also provide clues to a glaucoma diagnosis (e.g., facial features suggesting metabolic disorders, connective tissue disorders, chromosomal abnormalities). Occasionally, the ophthalmologist may be the first to suspect the systemic condition related to the ocular abnormality being examined (e.g., oculocerebrorenal or Lowe syndrome, neurofibromatosis).
Corneal Examination
This portion of the examination assesses the cornea for glaucoma-induced changes such as enlargement, edema, and scarring. Other abnormalities, if present, may also suggest coexisting ocular abnormalities (as with developmental glaucomas such as Axenfeld–Rieger or Peters, as discussed in Chapter 14).
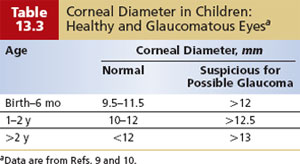
The healthy newborn’s cornea has a horizontal diameter ranging from 9.5 to 10.5 mm, which enlarges by about 0.5 to 1.0 mm in the first year of life (9–11) (Table 13.3). Distention of the globe in response to elevated IOP (buphthalmos) leads to enlargement of the cornea, especially at the corneoscleral junction. A corneal diameter larger than 12 mm in the first year of life is a highly suspect finding. Asymmetry in diameter between the two corneas or a corneal diameter of 13 mm or more at any age strongly suggests abnormality (3). Corneal enlargement is more obvious in asymmetric cases (Fig. 13.1). Simple inspection of the corneas will often identify asymmetric corneal diameters of as little as 0.25 mm, likely because of the examiner’s assessment of corneal area (rather than its diameter) by visual inspection. Corneal diameters can be measured by using a millimeter ruler held in the frontal plane in the office, or by calipers in the anesthetized state.
Acute, severe IOP elevation produces corneal enlargement in the newborn or infant, frequently accompanied by tears in the Descemet membrane (Haab striae). These often appear acutely as areas of increased corneal edema and clouding (3) (Figs. 13.5 and 13.6). In more advanced cases, dense opacification of the corneal stroma may persist despite IOP reduction (Fig. 13.10). In contrast, moderate IOP elevation insufficient to produce noticeable corneal opacity gradually enlarges the infant’s corneas, sometimes proceeding unnoticed if symmetric, while concurrent optic nerve damage progresses to severe degrees (Fig. 13.8).
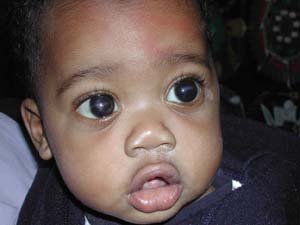
Figure 13.10 A 9-month-old boy with congenital glaucoma and residual corneal scarring from Haab striae, despite successful IOP control with angle surgery.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


