Glaucomas Associated with Disorders of the Corneal Endothelium
In the clinical setting of glaucoma and disorders of the cornea, it is helpful to consider two categories of conditions. In the first category, there are developmental anterior segment abnormalities, acquired conditions of the cornea, or primary disorders of the corneal endothelium that occur with glaucoma. In the second category, the cornea changes are secondary to the underlying glaucoma condition. Table 16.1 summarizes these categories and the various clinical disease categories. The subject of this chapter is primary corneal endothelial disorders that are associated with glaucoma, which include iridocorneal endothelial (ICE) syndrome; posterior polymorphous corneal dystrophy (PPCD), also called posterior polymorphic dystrophy (of cornea) (PPMD); and Fuchs endothelial dystrophy. (The remaining clinical conditions are discussed in other chapters.)

IRIDOCORNEAL ENDOTHELIAL SYNDROME
General Clinical Features and Terminology
The ICE syndrome is characterized by a primary corneal endothelial abnormality (1). Historically, three clinical variants were distinguished on the basis of changes in the iris (Table 16.2), but it is now recognized that progressive iris atrophy, Chandler syndrome, and Cogan–Reese syndrome represent a spectrum of ICE syndrome rather than distinct clinical identities. During the lifetime of a patient with ICE syndrome, the difference in clinical features may reflect the time at which the patient is seen. For example, a patient with initial findings of Chandler syndrome may later develop iris hole formation or nodules, changing the diagnosis to progressive iris atrophy or the Cogan–Reese syndrome, respectively. In other cases, however, the disease does not progress. Among the three clinical variants of ICE syndrome, Chandler syndrome appears to be the most common (2).

In general, the clinical features include presentation in early to middle adulthood, predilection for women, reduced visual acuity, pain, abnormalities of the iris, unilateral occurrence with variable amount of cornea edema but with subclinical abnormalities of the corneal endothelium in the fellow eye, anterior chamber angle abnormalities, and glaucoma (3,4). Familial cases are rare, and there is no consistent association with systemic diseases. In some cases, the corneal edema may occur at intraocular pressure (IOP) levels that are normal or only slightly elevated.
In one study of 37 consecutive cases of the ICE syndrome in the United States, Chandler syndrome was the most common clinical variation, accounting for 21 cases (56%), and was characterized by more severe corneal edema despite less severe glaucoma than in the rest of the group (2). However, in a study of 60 consecutive patients with ICE syndrome in Thailand, 38 patients had Cogan–Reese syndrome, 14 had Chandler syndrome, and 8 had progressive iris atrophy (5). In both studies, glaucoma occurred, most commonly in patients with progressive iris atrophy.
Pathologic Features
Changes in the Cornea
The common feature of the ICE syndrome is a corneal endothelial abnormality, which may be seen by slitlamp biomicroscopy as a “fine hammered silver” appearance of the posterior cornea, similar to that of Fuchs endothelial dystrophy (Fig. 16.1).

Figure 16.1 Slitlamp view shows the fine, beaten-silver appearance of a corneal endothelial abnormality in a patient with ICE syndrome.
The findings seen on specular microscopy of the corneal endothelium are virtually pathognomonic of the ICE syndrome. The affected endothelial cells appear dark by specular microscopy except for a light central spot and a light peripheral zone with various degrees of pleomorphism in size and shape and loss of the clear hexagonal margins (Fig. 16.2) (4,6). As discussed earlier under the clinical features, most patients who have ICE syndrome are symptomatic in one eye; however, abnormalities in the contralateral asymptomatic eye can be identified by specular microscopy.
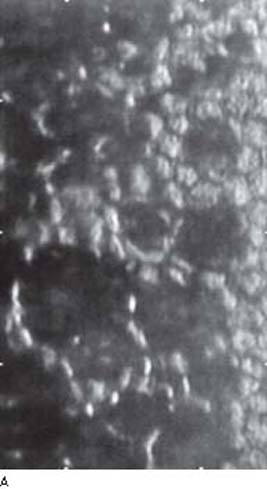

Figure 16.2 A: Specular microscopy of corneal endothelium in ICE syndrome. Cell borders are obscured, resulting in loss of the normal endothelial mosaic. Note dark areas within endothelial cells. Brighter reflections are believed to be from cell borders.B: Cornea, ICE syndrome. Scanning electron microscopy demonstrates sharp demarcation between abnormal (ICE) cells with microvilli and relatively unaffected endothelial cells. (From Cockerham GC, Kenyon KR. The corneal dystrophies. In: Tasman W, Jaeger EA, eds. Duane’s Clinical Ophthalmology. Vol 4. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:chap 16.)
Light microscopy of the cornea of a patient with ICE syndrome has revealed a monolayer of reduced cell density with occasional acellular zones and multiple endothelial layers, suggesting loss of contact inhibition (7). The ICE cells undergo metaplasia taking on morphologic features of epithelial cells, such as positive immunohistochemical staining for cytokeratins that are normally expressed by epithelium rather than endothelium (8). Some specimens have shown mononuclear inflammatory cells between the ICE cells (9), which support a proposed mechanism of viral-induced pathogenesis of ICE syndrome (as discussed later) (10).
This metaplasia is supported by scanning electron microscopy where the ICE cells showed numerous microvilli on the apical surface, in addition to cytoplasmic tonofilaments, and indentations on the basal surface containing clumps of fibrillar collagenous material (9). Other endothelial cells showed filopodium and cytoplasmic actin filaments suggesting migration (11). Another observation was a multilayered collagenous membrane posterior to the normal prenatal and postnatal layers of the collagenous structures of the Descemet membrane (12).
Changes in the Iris
The abnormalities of the iris constitute the primary basis for initially distinguishing clinical variations within the ICE syndrome, but as mentioned earlier, it is now recognized that these represent a spectrum the disease (3). In progressive iris atrophy, the hallmark is hole formation associated with corectopia and ectropion uvea that usually occur in the direction toward the quadrant with the most prominent area of peripheral anterior synechia (Fig. 16.3) (13). There appear to be two forms of atrophic iris holes. With stretch holes, the iris is markedly thinned in the quadrant away from the direction of pupillary distortion, and the holes develop within the area that is being stretched. In other eyes, melting holes develop without associated corectopia or thinning of the iris, which is thought to occur due to ischemia of the iris based on iris angiography (14).
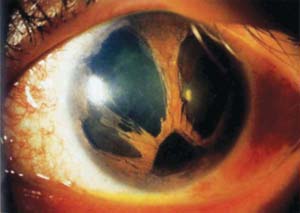
Figure 16.3 Eye of a patient with progressive iris atrophy, a variation of ICE syndrome, with extensive hole formation in the iris Patients typically have more marked corectopia (the pupil is in the inferior quadrant in this eye).
In Chandler syndrome, there may be no clinically appreciated iris changes or there is minimal corectopia and mild atrophy of the stroma of the iris (Fig. 16.4). In the Cogan–Reese syndrome, the iris is distinguished by pigmented, pedunculated nodules on the surface (Fig. 16.5).
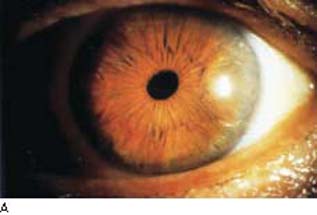
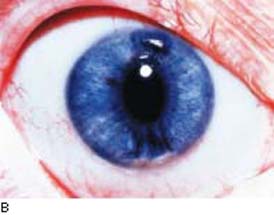
Figure 16.4 In patients with Chandler syndrome, a clinical variation of ICE syndrome, the eye may have a grossly normal-appearing iris or minimal corectopia and mild peripheral iris stromal atrophy (A), or more obvious iris changes, with a distorted, displaced pupil and variable degrees of iris stromal atrophy but no hole formation in the iris (B).
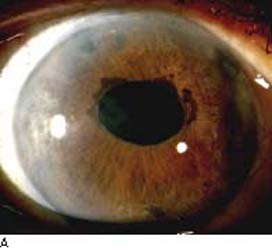
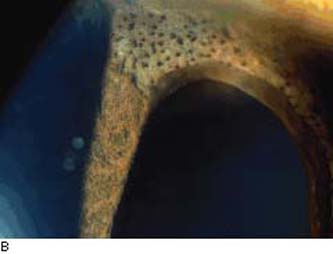
Figure 16.5 A: Cogan–Reese syndrome, a variation of ICE syndrome, shows ectropion uvea, some pupil distortion, and numerous dark nodules that are most prominent on the superior area of the iris stroma.B: ICE syndrome. Iris distortion and dark nodules (nevi) are present. Abnormal endothelium is often present on the iris surface. (From Cockerham GC, Kenyon KR. The corneal dystrophies. In: Tasman W, Jaeger EA, eds. Duane’s Clinical Ophthalmology. Vol 4. Philadelphia, PA: Lippincott Williams & Wilkins; 2006:chap 16.)
Light microscopy of the iris in ICE syndrome shows a cellular membrane on the anterior surface of the iris (Fig. 16.6), which is also referred to as a retrocorneal membrane that is continuous with that seen over the anterior chamber angle in the quadrant toward which the pupil is distorted (1,13). The nodular lesions that are characteristic of Cogan–Reese syndrome have an ultrastructure similar to that of the underlying stroma of the iris and are always surrounded by the previously described cellular membrane (15) (Fig. 16.7).
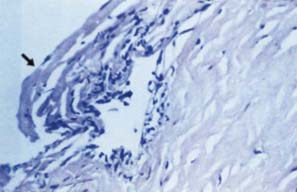
Figure 16.6 The glaucoma in ICE syndrome does not correlate precisely with the degree of synechial closure, and cases have been reported in which the angle was entirely open. In such cases, it is presumed that the trabecular meshwork is covered by a cellular membrane, consisting of a single layer of endothelial cells and Descemet-like membrane (arrow).
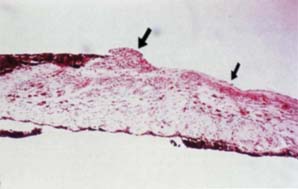
Figure 16.7 Light microscopic view of an iris specimen from an eye with Cogan–Reese syndrome.
Changes in the Lens
In rare cases, the retrocorneal membrane of the ICE syndrome may grow over the anterior lens surface, simulating the anterior lens capsule, which can create confusion when performing a capsulorrhexis during cataract surgery (16). This retrocorneal membrane can also appear on the anterior surface of an intraocular lens implant (Fig. 16.8).
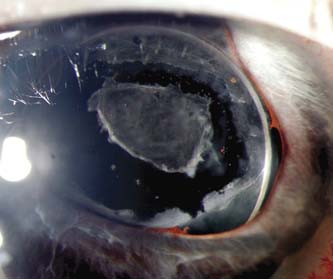
Figure 16.8 Retrocorneal membrane in ICE syndrome on the anterior surface of the intraocular lens, which was treated with an Nd:YAG anterior capsulotomy.
Gonioscopic Findings
Peripheral anterior synechia, usually extending to or beyond the Schwalbe line, is another clinical feature common to the ICE syndrome (Fig. 16.9). Elevated IOP usually begins as the synechiae progressively close the anterior chamber angle. However, the glaucoma does not correlate precisely with the degree of synechial closure and has been reported to occur when the entire angle was open but apparently covered by the cellular membrane (17,18).


Figure 16.9 A: Glaucoma occurs in a large proportion of patients with the ICE syndrome. In most cases, the glaucoma is associated with peripheral anterior synechiae, which usually extend to or beyond the Schwalbe line.B: This gonioscopic image shows the high peripheral anterior synechiae, especially in the center of the view, which also reveals a broad area of ectropion uveae. (Courtesy of William A. MacIlwaine IV, MD.)
These angle findings were confirmed in a histology study that revealed a Descemet-like membrane with a single layer of endothelial cells extending from the peripheral cornea and covering an open anterior chamber angle in some areas or synechial closure of the angle elsewhere in the same eye (1).
Theories of Mechanism
On the basis of the clinical and histopathologic evidence discussed earlier, Campbell and colleagues (13) proposed a “membrane theory” that a primary abnormality of the corneal endothelium was responsible for the findings in ICE syndrome (Fig. 16.10). The etiology that leads to the corneal endothelial changes is unknown. The absence of a positive family history and the presence of the postnatal layer of Descemet membrane suggest that it is an acquired disorder.
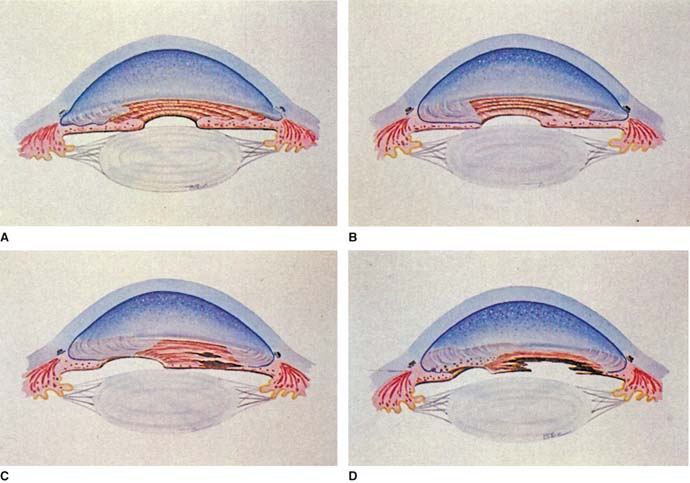
Figure 16.10 Membrane theory of Campbell for pathogenesis of ICE syndrome A: Extension of the membrane from the corneal endothelium over the anterior chamber angle and onto the iris. B: Contraction of the membrane, creating peripheral anterior synechiae and corectopia. C: Thinning and atrophy of iris in quadrants away from the corectopia. D: Hole formation in an area of atrophy (in progressive iris atrophy), ectropion uvea in the direction of the corectopia, and nodules in the area of the membrane (in Cogan–Reese syndrome). (From Shields MB. Progressive essential iris atrophy, Chandler syndrome, and the iris nevus [Cogan–Reese] syndrome: a spectrum of disease. Surv Ophthalmol. 1979;24(1):3–20, with permission.)
With evidence of inflammation in some cornea histopathology specimens, a virus-mediated mechanism has been proposed (7). Another study from this group supports this virus etiology for ICE syndrome based on the presence of DNA polymerase gene products for herpes simplex that was confirmed by sequencing the product (10). Specifically, among 31 cases (25 cornea specimens from patients with ICE syndrome and 6 from patients with chronic herpetic keratitis), 16 ICE syndrome (64%) and 4 herpetic keratitis (67%) were positive for herpes simplex virus DNA, which was localized to expression in the endothelium. Control corneas (n = 15) were negative for herpes simplex. In a smaller subset of nine ICE syndrome cases, the specimens were negative for the Epstein–Barr and herpes zoster viruses. With this high (64%), specific expression of herpes simplex (i.e., negative for Epstein–Barr and zoster viruses) in the cornea endothelium of ICE syndrome specimens, it is conceivable to consider that a herpes simplex–mediated infection of the cornea endothelium transforms these cells to lose contact inhibition and to transform into an epithelial-like cell. These biologically transformed cells form a proliferative cellular membrane across the anterior chamber angle, which can obstruct the trabecular meshwork, and onto the surface of the iris, which can lead to formation of peripheral anterior synechiae and to the various iris changes observed in the spectrum of progressive iris atrophy, Cogan–Reese syndrome, and Chandler syndrome.
Differential Diagnosis
There are several disorders of the cornea or iris, many of which have associated glaucoma, which can be confused with ICE syndrome. It is helpful to think of these in the following three categories: (a) endothelial disorders, (b) dissolution of the iris, and (c) nodular lesions of the iris.
Among the other corneal endothelial disorders, PPMD may be associated with glaucoma and changes of the anterior chamber angle and iris that resemble the ICE syndrome. However, the corneal abnormalities and other clinical features clearly distinguish these two spectra of disease (discussed in the next section). Specular microscopy may be helpful in distinguishing between ICE syndrome and PPMD (19). Fuchs endothelial dystrophy has changes in the cornea endothelium that are very similar to those of the ICE syndrome but none of the chamber angle or iris features of the latter condition (discussed in the last section).
In the category of dissolution of the iris, the Axenfeld–Rieger syndrome has striking clinical and histopathologic similarities to the ICE syndrome, but the congenital nature, bilaterality, and other features described in Chapter 14 help to separate the two conditions. Some advanced cases of progressive iris atrophy might resemble aniridia, but the bilaterality of the latter disorder is a helpful differential feature (Chapter 14). Iridoschisis is characterized by separation of superficial layers of iris stroma and may be associated with glaucoma, but it is typically a disease affecting older adults (Chapter 17).
Among the category of nodular lesions of the iris, melanomas of the iris (20); iris melanosis, which may be familial (21); Lisch nodules seen in neurofibromatosis type 1 (22); bilateral diffuse iris nodular nevi (23); and nodular inflammatory disorders, such as sarcoidosis, may also have pedunculated nodules strikingly similar to those of the Cogan–Reese syndrome.
Management
Patients with ICE syndrome may require treatment for corneal edema, the associated glaucoma, or both during the course of the disease. Given the unpredictable biological behavior of the abnormal corneal endothelium, patients with ICE syndrome should be monitored regularly based on signs and symptoms of the disease. For instance, one case has been reported in which the pigmented nodules appeared on the iris 20 years after the initial diagnosis of ICE syndrome (24). Although specular microscopy can help to diagnose ICE syndrome, the findings do not correlate with the degree of corneal edema or decompensation or with the level of IOP elevation (25).
In the early stages, the glaucoma can often be controlled medically, especially with drugs that reduce aqueous production. When the IOP can no longer be controlled medically, surgical intervention is indicated, and a high percentage of patients with the ICE syndrome eventually require surgery. Given the membrane theory of this disease (Fig. 16.10), laser trabeculoplasty is not effective for this disease and is not recommended as treatment. In one study of 66 patients, glaucoma occurred in 33 (50%), and 22 (66%) of these underwent surgery; 45% of the patients who had a trabeculectomy required more than one procedure (26). Filtering surgery is reasonably successful, although late failures have occurred because of endothelialization of the filtering bleb (27). Adjunctive use of 5-fluorouracil did not improve the surgical outcomes (28). Adjunctive mitomycin C has been reported to offer reasonable intermediate-term success (29). Another surgical option is glaucoma drainage-device surgery (30). However, repeated filtering procedures, glaucoma drainage-device revision, and cyclodestructive laser may be additional options to lower IOP if previous surgical interventions have failed.
The corneal edema may be improved by lowering the IOP, although the additional use of hypertonic saline solutions may be required. However, in corneas with marked dysfunction of the endothelium, the edema will not clear, and penetrating keratoplasty is usually indicated for this situation after the glaucoma has been controlled (31). In one series of 14 patients who had ICE syndrome treated by penetrating keratoplasty, repeated corneal grafts were required in 6 (43%) over an average follow-up of 58 months (32). In the future, the development of therapies can be directed more specifically at the underlying disease process. For example, if the theory of a viral cause proves to be correct, it may allow treatment with antiviral agents. Another approach may be to prevent the growth of the endothelial membrane. An immunotoxin has been described that inhibits the proliferation of human corneal endothelium in tissue culture.
POSTERIOR POLYMORPHOUS CORNEAL DYSTROPHY
General Clinical Features and Terminology
Posterior polymorphous corneal dystrophy is a rare, bilateral, autosomal dominant familial disorder of the corneal endothelium. Presently, the three genes that have been identified for PPMD are COL8A2 on chromosome 1p34.3 (33), ZEB1 (formerly called TCF8) on chromosome 10p11.2 (34), and VSX1 on chromosome 20p (35) (Chapter 8).
In general, the clinical features of PPMD include symptoms presenting in young adulthood and a clinical spectrum of characteristic corneal changes, peripheral iridocorneal adhesions, iris atrophy, and corectopia (discussed in detail later). Glaucoma occurs in approximately 15% of patients with PPMD (36).
Pathologic Features
Changes in the Cornea
By slitlamp biomicroscopy, the posterior cornea has the appearance of blisters or vesicles at the level of Descemet membrane (Fig. 16.11A). The vesicles may be linear (Fig. 16.11B) or in groups and may be surrounded by an aureole of gray haze (36). Band-like thickenings may also be seen at the level of Descemet membrane (37). On specular microscopy, several abnormal patterns have been described: either vesicles and band patterns at the level of Descemet membrane or a geographic pattern with associated haze of Descemet membrane and deep corneal stroma (38). An interesting observation was that 48 patients with PPMD, who had classic vesicles alone (42%) or vesicles with bands (48%) or diffuse abnormality of Descemet membrane (10%), had no other ocular abnormalities other than those of the cornea (39). In contrast, the latter specular pattern of prominent haze of Descemet membrane appears to be associated more with iridocorneal adhesions and glaucoma (38).
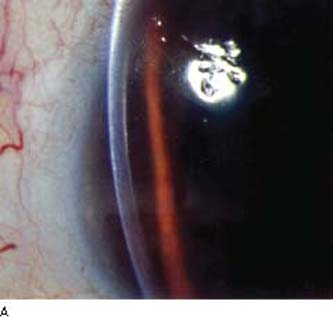
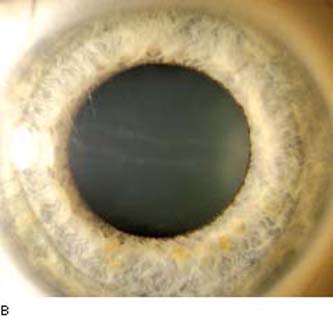
Figure 16.11 Slitlamp image of a patient with PPMD shows typical, irregular vesicular lesion of the posterior corneal surface (A) and subtle snail track across the central cornea (B).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


