11
Episcleritis, Scleritis, and Keratitis
Deanne Nakamoto and Paul A. Gaudio
This chapter aims to serve as a primer for diagnosing and treating patients who present with episcleritis, scleritis, or keratitis. In managing the inflammation of the eyewall—meaning scleritis, episcleritis, and keratitis—we follow the general treatment outline of (i) achieving immediate control of the inflammation and (ii) planning for long-term control, while (iii) preventing treatment toxicity, and (iv) optimizing the visual outcome. Corticosteroids and immunomodulatory drugs are the mainstay of treatment of these diseases.
EPISCLERITIS
Episcleritis is a bothersome but relatively benign inflammation of the episclera—the fibrous coat of tissue beneath the bulbar conjunctiva that overlies the sclera, generally synonymous with Tenon capsule. The episclera inserts posteriorly at the optic nerve and anteriorly 2 mm posterior to the limbus.
Presentation
- Bright and sectoral redness overlies the sclera, which often blanches with the application of topical phenylephrine. Hyperemic vessels are mobile over the sclera (unlike in scleritis) and generally are oriented radially. Notably, the palpebral conjunctiva is not involved, a finding that is useful in distinguishing episcleritis from conjunctivitis.
- Unlike scleritis, the inflamed area is not tender.
- Vision is unaffected.
- There are a few distinctive varieties of episcleritis:
- Nodular episcleritis is used when the inflamed area appears to cluster around discrete nodules.
- Diffuse episcleritis is used for any nonnodular presentation, even if it is localized to one or a few clock hours. This encompasses most cases of episcleritis.
- Pingueculitis is used when the inflammation is centered on a pinguecula—this entity may represent a different pathophysiologic process than true episcleritis.
- Orbital myositis is a less common variant; it is the inflammation of the anterior portion of a rectus muscle at its insertion to the sclera. Patients will have localized redness and will note pain when the muscle contracts or is put on stretch.
- Nodular episcleritis is used when the inflamed area appears to cluster around discrete nodules.
Workup
- Most episcleritis does not require a workup.
- Gather a careful review of systems (ROS) to rule out the very slight possibility that a patient’s episcleritis represents a forme fruste systemic autoimmune disease.
- If ROS is positive for joint pains, morning stiffness, rashes, nasopharyngeal ulcers, and vitiligo or if there is a strong family history of autoimmune disease, consider ordering a workup similar to that of scleritis (see later discussion).
Treatment of Episcleritis in the Absence of a Known Systemic Illness
In the absence of a known systemic illness, episcleritis is generally self-limited. Once the acute presentation has passed, it will subside, although recurrences are not uncommon. For this reason, episcleritis can often be treated acutely without the need for long-term control. Consider the following treatment options.
No Therapy
- Episcleritis occurring in the absence of a systemic disease never actually requires treatment since it is not visually disabling and usually will resolve if observed with sufficient patience.
Topical Therapy
- We start with artificial tears four times per day.
- We avoid corticosteroids in treating episcleritis. While these agents, given topically, invariably effect improvement in immune mediated episcleritis, all uveitis practitioners are familiar with patients who use topical steroids for immediate control of this condition, and are then unable to taper and discontinue the drug, ending up interminably dependent, often with unfortunate consequences. To our knowledge, topical nonsteroidal agents are ineffective for episcleritis.
Systemic Therapy
- Oral nonsteroidal anti-inflammatory drugs (NSAIDs) are near always effective in treating episcleritis, and we prefer this approach if one elects to treat at all. NSAIDs are grouped into several classes, and patients will often respond more favorably to one class of drugs than to another. Some common regimens are
- Indomethacin 150 mg per day, split into two or three doses
- Diflunisal 500 mg two times per day
- Tolmetin 1,200 mg per day, split into two or three doses or
- Naproxen 500 mg two times per day
Patients should take the drug with food and should not be taking corticosteroids orally, (the use of corticosteroids and NSAIDs concomitantly risks causing or exacerbating gastritis).
- Indomethacin 150 mg per day, split into two or three doses
- Reevaluate after 1 week.
- If the episcleritis is controlled at this point, then reduce the NSAID dose by one half or one third, and schedule follow-up in 1 to 2 weeks. At each follow-up visit, plan to taper the dose by one further increment if the disease remains controlled, until stopping the drug.
- If the disease is not controlled at the 1-week follow-up, change to a different NSAID, and schedule follow-up in 1 week, following the same tapering plan if the disease comes under control.
- Once an effective agent has been found, patients can often use the drug only as needed (e.g. for work or important occasions) rather than chronically. Eventually, the episcleritis will subside spontaneously and the drugs can be discontinued.
- If the episcleritis is controlled at this point, then reduce the NSAID dose by one half or one third, and schedule follow-up in 1 to 2 weeks. At each follow-up visit, plan to taper the dose by one further increment if the disease remains controlled, until stopping the drug.
Treatment of Episcleritis That Occurs with a Known Autoimmune Disease
If episcleritis occurs in the context of a known autoimmune illness, most commonly rheumatoid arthritis (RA) or lupus, then the treatment of the systemic illness needs to be increased until the episcleritis has resolved. Consider the episcleritis a sign that a patient’s systemic disease is not under adequate control.
Special Cases of Episcleritis
In some cases, the disease will not respond to NSAIDs, and if so, we consider the following special cases.
Herpes Infection
- Herpes simplex and herpes zoster can cause episcleritis, as well as keratitis or conjunctival ulceration. We suspect herpetic infection in patients who do not improve on NSAIDs or topical corticosteroids.
- Treat the suspected cases with acyclovir 400 mg five times per day, or equivalent doses of valacyclovir or famciclovir, adjusting as necessary if herpes zoster seems likely.
Chronic Episcleritis
- Very rarely, the episcleritis resists attempts to taper therapy and persists for many months. This unfortunate condition is a therapeutic dilemma. Our approach is to share this fact with the patient, reassure him or her that the condition will not hurt his or her vision, and then treat if necessary.
- Maintain vigilance for the development of a systemic disease, while checking autoimmune serologies every 6 months in these patients. Eventually, many of these patients turn out to have additional autoimmune problems in some form.
- Treatment is as for acute episcleritis, i.e., with cold artificial tears or oral nonsteroidal agents as needed.
- In our experience, the vast majority of chronic episcleritis is the result of having treated the condition initially with topical corticosteroids, and in this situation the condition will prove extremely difficult to treat if steroids are withdrawn. When patients present with this history, we do recommend discontinuing steroids and using oral NSAIDs instead to minimize the superficial inflammation, acknowledging that most patients will still sustain a prolonged period of eye redness—despite using oral NSAIDs—before the episcleritis finally subsides. When patients prove reticent to take this approach (which is common), the best one can do is to minimize the corticosteroid regimen in the hope of avoiding the worst long term side effects.
- Maintain vigilance for the development of a systemic disease, while checking autoimmune serologies every 6 months in these patients. Eventually, many of these patients turn out to have additional autoimmune problems in some form.
SCLERITIS
Scleritis is the inflammation of the sclera proper, associated with severe ocular as well as systemic morbidities. Patients with scleritis suffer not only from vision loss due to corneoscleral thinning most directly but also from uveitis, cataracts, and glaucoma. Recognizing and appropriately treating scleritis are important not only to prevent such ocular morbidity but also to help diagnose and aid in the management of systemic autoimmune disease. Necrotizing scleritis is largely associated with systemic diseases, while diffuse scleritis, nodular scleritis, and posterior scleritis are only sometimes caused by underlying systemic diseases. Scleritis is most commonly found in patients with RA, Wegener granulomatosis (WG), systemic lupus erythematosus (SLE), relapsing polychondritis, inflammatory bowel disease, sarcoidosis, polyarteritis nodosa (PAN), and infectious diseases such as syphilis, tuberculosis, and herpes. Other immune diseases that have been linked to scleritis include Cogan syndrome, Takayasu disease, and Churg-Strauss syndrome. Nonimmune causes of scleritis include bisphosphonate use, trauma, and intraocular tumors.
Scleritis affects women twice as often as men, and it is rare in children and the elderly. It usually affects patients between the ages of 30 and 50 with a peak in the fifth decade. Scleritis is bilateral in about 50% of patients.
Presentation
Scleritis may affect the anterior sclera, such that the redness is visible on examination, or the posterior sclera, where little or no redness is visible beneath the eyelids.
Anterior Scleritis
- Pain is the leading feature of scleritis. A typical patient will be found shielding the eye, noting deep and penetrating pain and tenderness. The tenderness will be immediately evident upon examination as well.
- The eye is red with a slightly bluish tinge, a feature which is best seen grossly (without a slit lamp) and optimally with natural light.
- Very severe scleritis involves tissue necrosis, resulting from the occlusion of the inflamed scleral vasculature. The subsequently infarcted tissue looks like a yellow-white, cheesy plaque beneath the conjunctiva.
- When scleritis resolves, it leaves behind a bluish tinge to the sclera, which is due to the collagen rearrangement thinning of the scleral fibers.
- There may be an adjacent corneal infiltration, in which case the condition is known as sclerokeratitis. Roughly 30% of anterior scleritis patients will have anterior chamber inflammation as well.
- One generally speaks of four types of anterior scleritis:
- Nodular scleritis. Roughly 20% of cases present with pain and discoloration centered on one or two raised nodules—hence the term for this presentation. The nodules are usually 3 to 6 mm in size, usually within a few millimeters of the limbus (Fig. 11.1).
- Diffuse scleritis. This is any scleritis that is not nodular, and it is the most common type of scleritis, representing about 60% of cases. It may involve between 1 and 12 clock hours of anterior sclera, usually contiguously. The affected area will bear the characteristic magenta hue and will often look slightly swollen compared to the surrounding tissue. This swelling is, in fact, fluid in the episcleral space, and if the process extends far enough posteriorly, it will be possible to image this fluid space with B-scan ultrasound (Fig. 11.2).
- Necrotizing scleritis with inflammation. This is the term used when the area of inflamed sclera includes necrotic tissue. It presents as a white mucus-like accumulation beneath the conjunctiva, surrounded by scleral injection. This condition is among the most painful of eye diseases and represents the most severe form of scleritis. It is associated with systemic diseases 50% to 70% of the time, and more than half of cases require immunosuppressive treatment for control. Some necrotizing scleritis with inflammation presents following an eye surgery, a condition termed surgically induced necrotizing scleritis (SINS). We workup and treat this condition as with any necrotizing scleritis.
- Necrotizing scleritis without inflammation (scleromalacia perforans). This less common, but more insidious, form may thin the sclera without the pain typically associated with scleritis. Often, the entire globe develops a dark gray appearance due to thinning. Necrotic tissue is apparent only intermittently and is often not evident on examination. The intraocular pressure may be elevated due to shutdown of the episcleral aqueous channels. These patients almost invariably have underlying RA, and the occurrence of this form of scleritis indicates very severe disease requiring immunosuppressive therapy.
- Nodular scleritis. Roughly 20% of cases present with pain and discoloration centered on one or two raised nodules—hence the term for this presentation. The nodules are usually 3 to 6 mm in size, usually within a few millimeters of the limbus (Fig. 11.1).
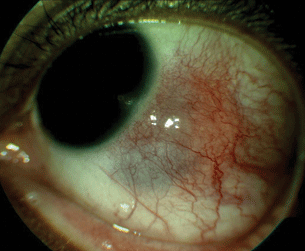
Figure 11.1 Nodular scleritis with sectoral inflammation of the sclera with elevated scleral nodule.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


