ANATOMY AND PHYSIOLOGY
Tear Film
The tear film is composed of three major layers, each produced by specialized cells. The adequacy and stability of the tear film are important in the maintenance of a healthy corneal surface. The normal pH of the tear film is 6.5 to 7.6. Tear film osmolarity is 296 to 308 mOsm (Figure 1.1; Table 1.1).
Conjunctiva
The conjunctiva is a nonkeratinizing stratified squamous epithelium with goblet cells. The conjunctiva extends from the limbus into the superior and inferior fornices to the gray line of the eyelid margin. The majority of goblet cells are concentrated in the fornices and at the caruncle. The conjunctiva is closely adherent to the superior and inferior tarsus by fine septa.
Cornea
The cornea has five distinct layers (Figure 1.2; Table 1.2). Maintenance of the orderly arrangement of the collagen fibers and the relative deturgescence of the corneal layers is important in permitting the cornea to remain optically clear.
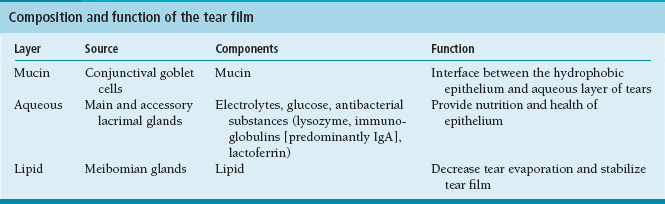
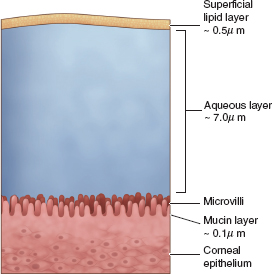
FIGURE 1.1. Structure of the tear film.
FIGURE 1.2. The five major layers of the cornea from anterior to posterior are the epithelium, Bowman layer, stroma, Descemet membrane, and endothelium.
The refractive index of the cornea is 1.376. To account for the combined optical power of the anterior and posterior curvatures of the cornea, a refractive index of 1.3375 is used to calibrate the keratometer.
The cornea is innervated by the long posterior ciliary nerves that branch from the ophthalmic divisions of cranial nerve V1.
Epithelium
The corneal epithelium is a stratified squamous epithelium that originates from limbal stem cells found in the palisades of Vogt and the interpalisade rete ridges. The epithelium has three predominant layers (Table 1.3).


Bowman Layer
Bowman layer or membrane is an acellular layer of compact collagen fibers. If damaged, Bowman’s does not regenerate, and scarring results.
Stroma
The thickest layer is the corneal stroma composed of type I collagen and glycosaminoglycans (mostly keratin sulfate) with scattered keratocytes. The lamellae are in orderly sheets that permit the dissection of a lamellar plane with a blunt instrument.
The imbibition pressure of the stroma = IOP–Swelling pressure.
Descemet Membrane
Descemet membrane is the basement membrane deposited by the endothelium.
 The anterior banded zone is formed before birth and is 3 to 4 mm thick.
The anterior banded zone is formed before birth and is 3 to 4 mm thick.
 The posterior nonbanded zone is continually added throughout life.
The posterior nonbanded zone is continually added throughout life.
 Guttae and Hassall-Henle bodies are focal excrescences (outgrowths) of Descemet membrane.
Guttae and Hassall-Henle bodies are focal excrescences (outgrowths) of Descemet membrane.
 In Fuchs dystrophy, central guttae precede stromal and epithelial edema and corneal decompensation.
In Fuchs dystrophy, central guttae precede stromal and epithelial edema and corneal decompensation.
Endothelium
The corneal endothelium is a monolayer of hexagonal cells that are not thought to divide or regenerate if damaged. At birth, the endothelium has 3,500 to 4,000 cells/mm2. With age, the cell count declines with 1,500 to 2,000 cells/mm2 present at adulthood. Corneal edema will occur when cells fall below 500 cells/mm2.
Sclera
 Made of type I collagen, proteoglycans and fibroblasts.
Made of type I collagen, proteoglycans and fibroblasts.
 Axenfeld loop is a branch of the posterior ciliary nerve that forms a visible intrascleral loop.
Axenfeld loop is a branch of the posterior ciliary nerve that forms a visible intrascleral loop.
 Senile scleral plaques are CaPO4 anterior to insertion of medial and lateral recti that become dehydrated leading to bluish color.
Senile scleral plaques are CaPO4 anterior to insertion of medial and lateral recti that become dehydrated leading to bluish color.
 Grayish scleral appearance can be due to scleral thinning allowing the underlying choroid to become visible.
Grayish scleral appearance can be due to scleral thinning allowing the underlying choroid to become visible.
Diagnostic Tests
The majority of corneal disease can be diagnosed using the slit lamp and careful examination; however, a number of diagnostic tests are available to help confirm the diagnosis or provide additional information. Below are listed some of the common tests and their applications (Table 1.4).
Anterior Blepharitis
Summary
A chronic inflammatory process of the eyelids, which may result secondarily in corneal changes.
Etiology
 Staphylococcal blepharitis is due to chronic colonization of the lid margins with staphylococcal species, marked by the presence of fibrin collarettes on the lashes.
Staphylococcal blepharitis is due to chronic colonization of the lid margins with staphylococcal species, marked by the presence of fibrin collarettes on the lashes.
 Seborrheic blepharitis is due to oily changes of the skin and is marked by greasy scales crusted on the lashes. These changes incite inflammatory processes in the eyelids. Inflammatory substances fall onto the ocular surface, where further inflammatory changes may occur. Seborrheic blepharitis is not characterized by corneal ulceration.
Seborrheic blepharitis is due to oily changes of the skin and is marked by greasy scales crusted on the lashes. These changes incite inflammatory processes in the eyelids. Inflammatory substances fall onto the ocular surface, where further inflammatory changes may occur. Seborrheic blepharitis is not characterized by corneal ulceration.
Signs and Symptoms
Patients complain of itching, irritation, tearing, or foreign body sensation, or a combination of these, with crusting on the lid margins and lashes.
Demographics
All races and ages may be affected.
Table 1.4 Diagnostic testing for corneal disease

DFA, direct fluorescent antibody; H&E, hematoxylin and eosin; HSV, herpes simplex virus; KOH, potassium hydroxide; PCR, polymerase chain reaction; PAS, periodic acid–Schiff; VZV, varicella–zoster virus.
Ophthalmic Findings
 Eyelids may be red and thickened and may show telangiectasias.
Eyelids may be red and thickened and may show telangiectasias.
 There may be ulceration and notching (ptilosis) of the lid margin (Figure 1.3).
There may be ulceration and notching (ptilosis) of the lid margin (Figure 1.3).
 Hordeolum and chalazion formation is seen more frequently.
Hordeolum and chalazion formation is seen more frequently.
 Lashes may be lost (madarosis) or misdirected.
Lashes may be lost (madarosis) or misdirected.
 Secondary corneal changes may be seen including pannus, punctate epithelial keratitis, and marginal infiltrates, because of a type IV hypersensitivity.
Secondary corneal changes may be seen including pannus, punctate epithelial keratitis, and marginal infiltrates, because of a type IV hypersensitivity.
Systemic Findings
 Seborrheic blepharitis is associated with rosacea.
Seborrheic blepharitis is associated with rosacea.
 Patients with seborrheic blepharitis may also have oily skin and dandruff.
Patients with seborrheic blepharitis may also have oily skin and dandruff.
Special Tests
Lid cultures should be taken periodically to guide antibiotic therapy.
Pathology
Chronic inflammation is present.
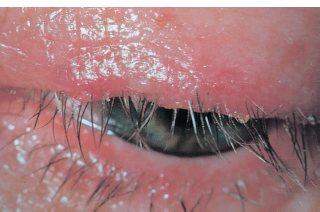
FIGURE 1.3. Crusting, lash loss, and ulceration of the lid margin caused by staphylococcal lid disease.
Disease Course
Chronic with exacerbations and remissions
Treatment and Management
Lid hygiene, consisting of warm-water soaks to loosen debris, followed by lid margin scrubs (with plain water, diluted baby shampoo, or an antibiotic drop) should be performed daily.
Medications
Antibiotics may be useful, guided by culture results. If chronic use is required, antibiotics should be rotated to minimize resistance.
Follow-Up
Patients often return frequently, since the problem is chronic. Often, no change in therapy is needed—just explanation, support, and empathy.
Differential Diagnosis
Unilateral disease is unusual, and a masquerade syndrome should be considered. Sebaceous gland carcinoma presents with unilateral eyelid ulceration, lid distortion, and lash loss. This requires a full-thickness biopsy and pathologic examination.
Posterior Blepharitis
Other names: meibomian gland dysfunction, meibomitis.
Summary
Meibomian gland dysfunction and inflammation of the meibomian glands change the composition of the lipid layer, resulting in instability of the tear film. Release of inflammatory mediators irritates the lid margin and ocular surface.
Etiology
Genetic predisposition in some patients, since seen more commonly in patients with rosacea
Signs and Symptoms
Burning and stinging.
Symptoms worse upon waking in the morning.
Redness and ocular irritation.
Sore eyelids.
Demographics
Very common, may be more frequent in patients with rosacea.
Ophthalmic Findings
 Foamy tears and oily tear film (Figure 1.4).
Foamy tears and oily tear film (Figure 1.4).
 Thickened and inspissated meibomian gland secretions.
Thickened and inspissated meibomian gland secretions.
 Stenotic or occluded meibomian gland orifices.
Stenotic or occluded meibomian gland orifices.
 Meibomian gland cysts and chalazia.
Meibomian gland cysts and chalazia.
 Telangiectasia of the lid margins.
Telangiectasia of the lid margins.
 Inferior interpalpebral staining with rose bengal or lissamine green.
Inferior interpalpebral staining with rose bengal or lissamine green.
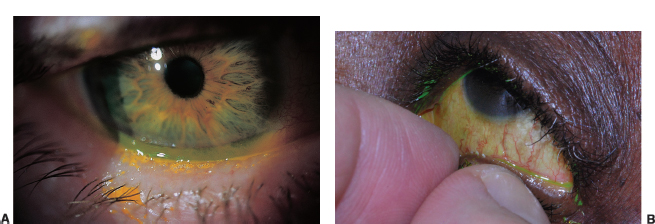
FIGURE 1.4. A: Foamy tears along the lower lid from saponification of the lipid layer in meibomian gland dysfunction. B: Inspissation of meibomian glands in posterior blepharitis.
Systemic Findings
Telangiectasia on the cheeks and nose.
Rosacea.
Disease Course
 Chronic disease with episodes of exacerbation.
Chronic disease with episodes of exacerbation.
 If severe, can lead to corneal neovascularization.
If severe, can lead to corneal neovascularization.
Treatment and Management
 Hot compresses.
Hot compresses.
 Oral doxycycline or tetracyclines.
Oral doxycycline or tetracyclines.
 Topical steroids and antibiotic.
Topical steroids and antibiotic.
 Artificial tears.
Artificial tears.
Differential Diagnosis
 Staphylococcal blepharitis.
Staphylococcal blepharitis.
 Keratoconjunctivitis sicca (KCS).
Keratoconjunctivitis sicca (KCS).
Hordeolum
Summary
An acute focal swelling of the eyelid secondary to obstruction of a sebaceous gland orifice (usually a meibomian gland, but may be a gland of Zeis or lash follicles). Internal hordeola occur at the posterior eyelid from meibomian gland inspissation. External hordeola, or styes, occur on the anterior eyelid in the glands of Zeis or lash follicles.
Etiology
Underlying meibomitis is usually present, with thickening of the gland secretions, stasis of oily gland secretions, and inspissation of the orifice. Staphylococcal infection is often present secondarily.
Signs and Symptoms
Patients present with an eyelid lump or swelling and erythema, which is often tender. There may be blurring of vision secondary to induced astigmatism and an oily tear film.
Demographics
All individuals may be affected.
Ophthalmic Findings
Lid tenderness, erythema, swelling, and focal mass. The blocked meibomian gland orifice is usually visible.
Systemic Findings
None.
Special Tests
None.
Pathology
Inflammation is caused as the oily material extends from the blocked gland into the surrounding tissue. A chalazion is present when lipogranulomatous inflammation occurs in response to lipids from meibomian or Zeis glands.
Disease Course
Generally self-limited as the lesion spontaneously drains. Granuloma formation may develop if the hordeolum does not drain, and a chalazion results, which is a nontender and uninflamed nodule.
Treatment and Management
 Frequent warm compresses at onset encourage spontaneous drainage.
Frequent warm compresses at onset encourage spontaneous drainage.
 Incision and drainage may be required (usually for cosmetic purposes).
Incision and drainage may be required (usually for cosmetic purposes).
 Intralesional steroid injection.
Intralesional steroid injection.
Medications
A local superinfection may require topical (or rarely, oral) antibiotics.
Follow-Up
None required.
Differential Diagnosis
Preseptal cellulitis may present as a diffuse infectious lid inflammation with absence of a focal mass. There is usually a history of trauma as the inciting event for infection. Insect bites and contact dermatitis are usually discerned by history. Recurrent chalazia in the same location, particularly in an older individual, should raise the suspicion for sebaceous cell carcinoma and may require biopsy.
Contact Dermatitis
Summary
Rapid onset of periorbital erythema, with thickening and swelling of the eyelid skin.
Etiology
 Type IV hypersensitivity reaction to a topically applied substance, usually a medication.
Type IV hypersensitivity reaction to a topically applied substance, usually a medication.
 Chief offenders include atropine, pilocarpine, epinephrine, apraclonidine, brimonidine, prostaglandins, antivirals, and antibiotics, particularly neomycin and other aminoglycosides.
Chief offenders include atropine, pilocarpine, epinephrine, apraclonidine, brimonidine, prostaglandins, antivirals, and antibiotics, particularly neomycin and other aminoglycosides.
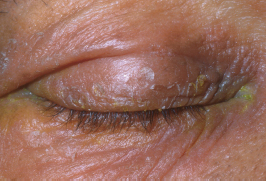
FIGURE 1.5. Severe red, scaly, and itchy periocular skin secondary to the topical use of medications such as neomycin ointment.
Signs and Symptoms
Periorbital redness, crusting, and itching. There may be a mild watery discharge.
Ophthalmic Findings
 Periorbital edema, erythema, and thickening.
Periorbital edema, erythema, and thickening.
 The skin may have a shiny, scaly, leathery appearance and texture (Figure 1.5).
The skin may have a shiny, scaly, leathery appearance and texture (Figure 1.5).
Systemic Findings
None.
Special Tests
None.
Pathology
Type IV allergic reaction, involving delayed type hypersensitivity mediated by T cells.
Disease Course
Chronic until the offending agent removed.
Treatment and Management
 Identification of the offending agent, which must be discontinued.
Identification of the offending agent, which must be discontinued.
 Cold compresses.
Cold compresses.
Medications
Oral antihistamines may help, as may application of a mild steroid preparation.
Follow-Up
Resolution should be apparent in approximately 2 weeks. Conservative measures should be used first, with addition of steroids at that time if necessary.
Differential Diagnosis
There is usually little confusion with this clinical appearance and supporting history of a recent addition of an offending agent.
Atopic Dermatitis
Summary
Chronic superficial inflammation of the skin, frequently associated with underlying manifestations of allergic eye disease (atopic keratoconjunctivitis).
Etiology
A form of type I hypersensitivity reaction involving immunoglobulin E (IgE) overproduction, eosinophils, and mast cells, with release of histamine and other inflammatory mediators.
Signs and Symptoms
Itching, burning, watering, and photophobia are prominent. Skin thickening and mucous discharge may be evident.
Demographics
Onset generally in the teens and early years of adulthood, extending until approximately age 60 years, at which time the disease process seems to burn itself out.
Ophthalmic Findings
 “Allergic shiners” or increased pigmentation of the periorbital skin (Figure 1.6).
“Allergic shiners” or increased pigmentation of the periorbital skin (Figure 1.6).
 Scaling and woody, thickened (often described as leathery or lichenified) eyelid skin, which may lead to ectropion, ptosis, and/or lagophthalmos with secondary ocular surface changes.
Scaling and woody, thickened (often described as leathery or lichenified) eyelid skin, which may lead to ectropion, ptosis, and/or lagophthalmos with secondary ocular surface changes.
 Inferior fornix subepithelial fibrosis.
Inferior fornix subepithelial fibrosis.
 Punctate epithelial keratitis and corneal epithelial defects, which may scar or develop microbial ulceration.
Punctate epithelial keratitis and corneal epithelial defects, which may scar or develop microbial ulceration.
 Associations with subcapsular cataracts, keratoconus, increased incidence of herpes simplex virus (HSV) eye disease (bilateral in some cases), and retinal detachment.
Associations with subcapsular cataracts, keratoconus, increased incidence of herpes simplex virus (HSV) eye disease (bilateral in some cases), and retinal detachment.
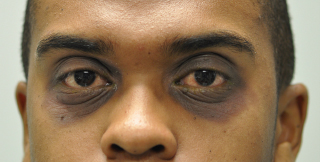
FIGURE 1.6. Periorbital skin pigmentation known
Systemic Findings
Asthma, atopic dermatitis or eczema, and allergic rhinitis or hay fever often coexist in the same individual and are frequently found in other family members.
Special Tests
Generally none. Rarely, a skin biopsy may be considered to assist with diagnosis. A conjunctival scraping may show eosinophils and mast cells.
Pathology
A prominence of mast cells and eosinophils is seen in the mucosal tissues.
Disease Course
Chronic, with exacerbations and remissions.
Treatment and Management
Any identifiable triggers should be avoided. Disease control is optimized by both local and systemic medications. Lid and tear abnormalities must also be addressed. Cold compresses.
Medications
 Artificial tears.
Artificial tears.
 Topical nonsteroidal anti-inflammatory drugs (NSAIDs), antihistamines, mast cell stabilizers (cromolyn, lodoxamide, olopatadine, bepotastine): may be continued indefinitely.
Topical nonsteroidal anti-inflammatory drugs (NSAIDs), antihistamines, mast cell stabilizers (cromolyn, lodoxamide, olopatadine, bepotastine): may be continued indefinitely.
 Steroids: should be the first drug to be discontinued.
Steroids: should be the first drug to be discontinued.
 Oral nonsedating antihistamines are a useful addition to therapy and may help to limit the number of topically applied agents (and preservative toxicity to a fragile ocular surface).
Oral nonsedating antihistamines are a useful addition to therapy and may help to limit the number of topically applied agents (and preservative toxicity to a fragile ocular surface).
 Cyclosporin has been used topically and systemically in severe cases.
Cyclosporin has been used topically and systemically in severe cases.
 Epithelial defects should be treated with prophylaxis against microbial infection with topical broad-spectrum antibiotics.
Epithelial defects should be treated with prophylaxis against microbial infection with topical broad-spectrum antibiotics.
Follow-Up
Regular intervals should be scheduled based on disease severity.
Differential Diagnosis
Vernal keratoconjunctivitis may present similar ocular findings, but the dermatitis is absent and generally affects young male patients.
Reference
Foster CS, Calonge M. Atopic keratoconjunctivitis. Ophthalmology 1990;97:992–1000.
Ichthyosis
Summary
Widespread abnormality of keratinization of the skin.
Etiology
Inherited. All inheritance patterns are seen, with autosomal dominant being most common and X-linked recessive the most severe.
Signs and Symptoms
Dry, scaly skin giving the appearance of a dirty neck, often referred to as the “dirty neck syndrome.”
Demographics
Becomes apparent in the first year of life.
Ophthalmic Findings
 Secondary lid abnormalities are seen, including ectropion, lagophthalmos, and resulting corneal exposure.
Secondary lid abnormalities are seen, including ectropion, lagophthalmos, and resulting corneal exposure.
 Pre-Descemet stromal deposits are seen, which are gray, punctate, and linear opacities without affecting vision.
Pre-Descemet stromal deposits are seen, which are gray, punctate, and linear opacities without affecting vision.
 Congenital cataracts may be present.
Congenital cataracts may be present.
 Prominent corneal nerves.
Prominent corneal nerves.
Systemic Findings
Skin changes as described above.
Special Tests
None.
Pathology
Abnormal keratinization of the skin.
Disease Course
Chronic.
Treatment and Management
Supportive measures for secondary ocular abnormalities. Not responsive to steroids.
Medications
None specific.
Follow-Up
As required depending on degree of ocular involvement.
Reference
Jay B, Blach RK, Wells RS. Ocular manifestations of ichthyosis. Br J Ophthalmol 1968;52:217–226.
Ectodermal Dysplasia
Summary
A group of congenital, nonprogressive abnormalities of epidermis and at least one of the following: hair, nails, teeth, sweat glands.
Etiology
Various inheritance patterns are seen.
Signs and Symptoms
Demographics
Congenital.
Ophthalmic Findings
 Blepharitis.
Blepharitis.
 Ankyloblepharon.
Ankyloblepharon.
 Hypoplastic lacrimal ducts.
Hypoplastic lacrimal ducts.
 Dry eye.
Dry eye.
 Corneal scarring and neovascularization.
Corneal scarring and neovascularization.
 Cataract.
Cataract.
 Glaucoma.
Glaucoma.
Systemic Findings
Skin changes as described above.
Special Tests
None.
Pathology
Reduction in number of sweat glands, sebaceous glands and hair follicles, depending on the type.
Disease Course
Children with anhidrotic ectodermal dysplasia cannot sweat properly, making hyperpyrexia a problem.
Treatment and Management
Supportive measures for secondary ocular abnormalities.
Medications
None specific.
Follow-Up
As required, depending on degree of ocular involvement.
Rosacea
Summary
Chronic disorder affecting the facial skin and eyes.
Etiology
Unknown. Multiple mechanisms have been implicated, including vascular changes of the skin, sunlight-induced damage, and type IV hypersensitivity reactions. Antigens including the human follicle mite Demodex have been implicated as inciting inflammatory agents.
Signs and Symptoms
Patients complain of ocular irritation, redness, foreign body sensation, and burning. Facial flushing, eyelid margin telangiectasias, erythema, pustules, papules, and rhinophyma may be noted.
Demographics
All ages, but generally aged 30 to 60 years. Rosacea is more common in whites and more frequently diagnosed in women, but the disease is more severe in affected male patients.
Ophthalmic Findings
 Lid telangiectasias.
Lid telangiectasias.
 Blepharitis.
Blepharitis.
 Meibomitis.
Meibomitis.
 Chalazia.
Chalazia.
 Conjunctival injection.
Conjunctival injection.
 Tear deficiency and instability.
Tear deficiency and instability.
 Corneal punctate epithelial keratitis.
Corneal punctate epithelial keratitis.
 Corneal vascularization.
Corneal vascularization.
 Inferior spade-shaped corneal infiltrates.
Inferior spade-shaped corneal infiltrates.
Systemic Findings
The skin of the face and upper body is affected. Facial flushing, telangiectasias (Figure 1.7), erythema, pustules, papules, and rhinophyma may be noted.
Special Tests
None.
Pathology
No unique features.
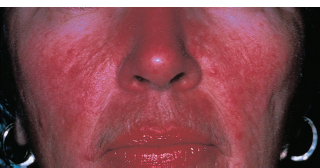
FIGURE 1.7. The typical rosacea facies with dilated, telangiectatic vessels on the cheeks and nose.
Disease Course
Chronic with exacerbations and remissions.
Treatment and Management
Local measures for lid hygiene, bacterial superinfection, and tear disturbances should be applied as required.
Medications
 Chronic use of oral tetracycline (doxycycline or minocycline) is advocated and may dramatically improve both skin and ocular disease.
Chronic use of oral tetracycline (doxycycline or minocycline) is advocated and may dramatically improve both skin and ocular disease.
 Topical metronidazole gel or cream has also been helpful.
Topical metronidazole gel or cream has also been helpful.
 Topical antibiotic use should be guided by culture results and rotated if used long term.
Topical antibiotic use should be guided by culture results and rotated if used long term.
 Topical steroid can be used to decrease sterile inflammation.
Topical steroid can be used to decrease sterile inflammation.
Follow-Up
Regular follow-up intervals are warranted for secondary ocular changes and monitoring during therapy.
Differential Diagnosis
 Contact dermatitis.
Contact dermatitis.
 Systemic lupus erythematosus (SLE).
Systemic lupus erythematosus (SLE).
 Seborrheic dermatitis.
Seborrheic dermatitis.
 Acne vulgaris may give similar skin changes but usually is distinguished by history and clinical features.
Acne vulgaris may give similar skin changes but usually is distinguished by history and clinical features.
 Rhinophyma may rarely be confused with basal cell carcinoma, and biopsy undertaken if necessary.
Rhinophyma may rarely be confused with basal cell carcinoma, and biopsy undertaken if necessary.
Reference
Knox CM, Smolin G. Rosacea. Int Ophthalmol Clin 1997;37:29–40.
Anatomy
Follicles
Follicles are focal collections of lymphocytes in the substantia propria of the conjunctiva. Follicles are most prominent in the inferior fornix as gelatinous elevations 1 to 5 mm in size with blood vessels coursing around the base of the follicles.
Common Causes
 Viral infections (adenovirus, herpes, molluscum contagiosum).
Viral infections (adenovirus, herpes, molluscum contagiosum).
 Chlamydia (adult inclusion, trachoma).
Chlamydia (adult inclusion, trachoma).
 Drugs (atropine, epinephrine, glaucoma medications, trifluridine).
Drugs (atropine, epinephrine, glaucoma medications, trifluridine).
Papillae
Papillae represent a nonspecific response to conjunctival irritation. Because of inflammation, pinpoint blood vessels in the tarsal conjunctiva become dilated, leaking fluid and inflammatory cells. A papillary response can best be seen on the superior tarsal conjunctiva as multiple small bumps, each with a central core of vessels.
Common Causes
 Bacterial infection.
Bacterial infection.
 Allergy/Atopy.
Allergy/Atopy.
 Floppy eyelid syndrome.
Floppy eyelid syndrome.
Inflammation of any cause (eye rubbing, surface keratinization, etc.).
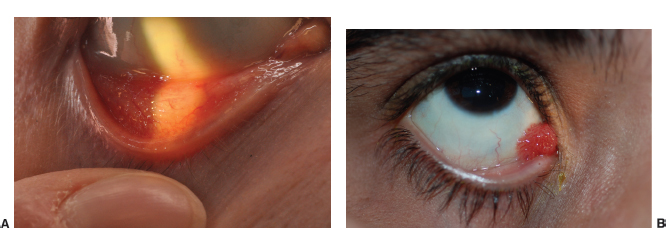
FIGURE 1.8. A: Follicles in the lower fornix with typical gelatinous, dome-shaped appearance. B: Papillae on the upper tarsal conjunctiva.
Giant Papillae and Cobblestone Papillae
The size of papillae is usually restricted by the fibrous septa attaching the conjunctiva to the underlying tarsus. With severe and chronic inflammation, these septa rupture, and the giant papillae coalesce to form the giant papillae. Stellate scarring can occur on the top of the papillae.
Common Causes
 Contact lens–associated giant papillary conjunctivitis (soft contact lenses more frequently).
Contact lens–associated giant papillary conjunctivitis (soft contact lenses more frequently).
 Vernal keratoconjunctivitis.
Vernal keratoconjunctivitis.
Membranes and Pseudomembranes
Damage to the conjunctival vessels and the epithelial surface by severe inflammation can cause exudation of fibrin, inflammatory cells, necrotic cells, and serous fluid. Coalescence of this material on the surface forms the membrane. Removal of a membrane causes bleeding from the raw epithelial surface; however, a pseudomembrane does not cause bleeding of the underlying surface. The differentiation of membranes from pseudomembranes is mostly of academic interest.
Common Causes
Severe epidemic keratoconjunctivitis (EKC).
Ligneous conjunctivitis.
Alkali or acid burns.
Stevens-Johnson syndrome (SJS).
Ocular cicatricial pemphigoid (OCP).
Gonococcal conjunctivitis.
Corynebacterium diphtheriae conjunctivitis.
Beta-hemolytic streptococcus conjunctivitis.
Allergic/Hypersensitive Conjunctivitis
Phlyctenule
Summary
A phlyctenule is a focal lymphocytic nodule usually at the limbus or on the bulbar conjunctiva that leads to localized fibrosis and vascularization. Phlyctenules are thought to be a type IV hypersensitivity reaction and can be a feature of staphylococcal blepharoconjunctivitis.
Etiology
The actual pathogenesis is unclear; however, phlyctenules are presumably a type IV hypersensitivity reaction to Staphylococcus organisms or tuberculosis (TB).
Signs and Symptoms
Foreign body sensation.
Redness.
Demographics
Any age group may be affected without sex predilection.
Ophthalmic Findings
 Unilateral.
Unilateral.
 Conjunctival injection.
Conjunctival injection.
 Pearly nodule at limbus or on bulbar conjunctiva.
Pearly nodule at limbus or on bulbar conjunctiva.
 Staphylococcal lid disease.
Staphylococcal lid disease.
 Inflammatory mound with ulcerated apex (Figure 1.9).
Inflammatory mound with ulcerated apex (Figure 1.9).
 Leash of vessels from conjunctiva.
Leash of vessels from conjunctiva.
Special Tests
 Lid cultures.
Lid cultures.
 Purified protein derivative (PPD) skin test, chest x-ray, if TB suspected.
Purified protein derivative (PPD) skin test, chest x-ray, if TB suspected.
Pathology
Focal lymphocytic nodules at limbus.
Disease Course
Fibrosis and vascularization of the nodule may occur, and a pannus may form at the peripheral cornea.
Treatment and Management
Direct treatment of the underlying cause (e.g., staphylococcal blepharitis).
Medications
Antibiotic ointment.
Low-dose steroid drops.
Differential Diagnosis
Pterygium.
Fuchs marginal keratitis.
Conjunctival intraepithelial neoplasia (CIN).
Conjunctival granuloma.
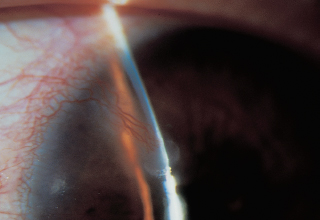
FIGURE 1.9. Elevated, ulcerated fibrovascular growth on the cornea in a 25-year-old patient with a phlyctenule.
References
Abu el-Asrar AM, Tabbara KF. Tetracycline treatment of phlyctenulosis. Ophthalmology 1994;101:1161–1162.
Ballintine EJ, Rotbart DC, Takimura Y. Phlyctenulosis. Am J Ophthalmol 1972;73:459–460.
Hay Fever (Allergic) Conjunctivitis
Summary
An immediate type I, IgE-mediated hypersensitivity reaction induced by airborne allergens. This occurs more frequently in patients with atopy or asthma.
Etiology
Type I hypersensitivity reaction to airborne allergens.
Signs and Symptoms
Severe itching.
Eyelid swelling.
Tearing.
Discharge.
Rapid development after exposure.
Ophthalmic Findings
Mucosal discharge.
Conjunctival hyperemia.
Papillary conjunctival reaction.
Conjunctival chemosis.
Eyelid swelling.
Caruncular hyperemia.
Systemic Findings
Rhinorrhea and sneezing.
Possibly asthma.
Special Tests
Conjunctival cytologic study with Wright stain shows eosinophils or granules.
Pathology
Seasonal allergic conjunctivitis is accompanied by increased mast cell numbers.
Disease Course
Seasonal exacerbation when exposed to allergen.
Treatment and Management
Cold compresses.
Vasoconstrictors.
Antihistamines.
NSAID.
Mast cell stabilizers.
Topical steroids.
Medications
See above.
Differential Diagnosis
Vernal conjunctivitis.
Medicamentosa.
Atopic keratoconjunctivitis.
Reference
Bonini S, Magrini L, Rotiroti G, et al. The eosinophil and the eye. Allergy 1997;52(Suppl):44–47.
Atopic Keratoconjunctivitis
Summary
Atopic keratoconjunctivitis is a chronic conjunctival inflammatory condition that occurs in patients with atopy, that is, three or more of the following features:
1. Typical, eczematoid skin lesions.
2. Pruritus.
3. Relapsing eczematoid dermatitis.
4. Personal or family history of asthma or allergic rhinitis.
Etiology
Hypersensitive immune system reacting to many antigens.
Signs and Symptoms
Itching.
Foreign body sensation.
Mucoid discharge.
Red eyes.
Ophthalmic Findings
 Periocular scaly skin and thickening of the eyelids: allergic shiners.
Periocular scaly skin and thickening of the eyelids: allergic shiners.
 Medium-sized papillae that can appear milky when confluent (Figure 1.10A).
Medium-sized papillae that can appear milky when confluent (Figure 1.10A).
 Conjunctival edema.
Conjunctival edema.
 Elevated limbal nodules: Horner-Trantas dots.
Elevated limbal nodules: Horner-Trantas dots.
 Extensive corneal neovascularization may occur.
Extensive corneal neovascularization may occur.
 Pseudogerontoxon (arc-shaped peripheral corneal opacity similar in appearance to a senile gerontoxon [Figure 1.10B]).
Pseudogerontoxon (arc-shaped peripheral corneal opacity similar in appearance to a senile gerontoxon [Figure 1.10B]).
 Conjunctival scarring (forniceal foreshortening and symblephera) may occur.
Conjunctival scarring (forniceal foreshortening and symblephera) may occur.
 Posterior subcapsular or shield-shaped anterior lens opacities.
Posterior subcapsular or shield-shaped anterior lens opacities.
Systemic Findings
 Skin lesions.
Skin lesions.
 Flexural lichenification in adults (eczema).
Flexural lichenification in adults (eczema).
 Facial and extensor involvement in infants or children.
Facial and extensor involvement in infants or children.
 Chronic relapsing dermatitis.
Chronic relapsing dermatitis.
 Asthma.
Asthma.
 Allergic rhinitis.
Allergic rhinitis.
 Family history of asthma or allergic rhinitis.
Family history of asthma or allergic rhinitis.
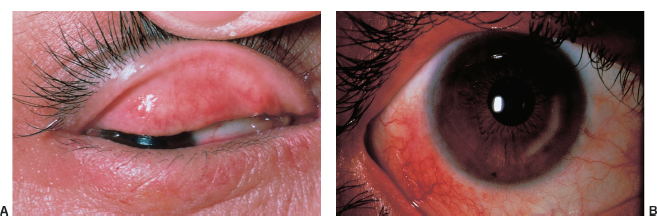
FIGURE 1.10. A: Confluent papillae and milky edema on the superior tarsus in an atopic patient. B: An arc-shaped deposition known as a pseudogerontoxon.
Special Tests
Conjunctival cytologic studies with eosinophils (less degranulated and less numerous than vernal).
Disease Course
 Chronic, progressive corneal pannus formation.
Chronic, progressive corneal pannus formation.
 Conjunctival scarring with long-standing disease.
Conjunctival scarring with long-standing disease.
 Bilateral ocular HSV infection may be seen.
Bilateral ocular HSV infection may be seen.
 Patients typically older than those with vernal keratoconjunctivitis.
Patients typically older than those with vernal keratoconjunctivitis.
Treatment and Management
 Removal of environmental triggers.
Removal of environmental triggers.
 Treatment is based on the severity of the condition and consists of topical vasoconstrictors, topical cromolyn sodium and other mast cell stabilizers, antihistamines, and NSAID drops.
Treatment is based on the severity of the condition and consists of topical vasoconstrictors, topical cromolyn sodium and other mast cell stabilizers, antihistamines, and NSAID drops.
 Short-term steroids or cyclosporin A for severe cases.
Short-term steroids or cyclosporin A for severe cases.
Medications
Disodium cromolyn.
Lodoxamide.
Levocabastine.
Epinastine
Bepotastine.
Ketotifen.
Olopatadine.
Oral antihistamines.
Topical or oral steroids.
Cyclosporin A: for refractory cases.
Differential Diagnosis
Vernal conjunctivitis.
Giant papillary conjunctivitis.
Hay fever.
Reference
Casey R, Abelson MB. Atopic keratoconjunctivitis. Int Ophthalmol Clin 1997;37:111–117.
Vernal Keratoconjunctivitis
Summary
Vernal keratoconjunctivitis is a seasonally recurring, bilateral inflammation of the conjunctiva that occurs predominantly in male children and young adults who often have a history of atopy. There are two forms, palpebral and limbal.
Etiology
Hypersensitivity to airborne allergens superimposed on an atopic predisposition.
Signs and Symptoms
Itching.
Photophobia.
Blurred vision.
Copious mucous discharge.
Demographics
Male children and young adults.
Hot, dry climates.
Ophthalmic Findings
 Blepharospasm and ptosis.
Blepharospasm and ptosis.
 Predominantly palpebral inflammation.
Predominantly palpebral inflammation.
 Diffuse papillary hypertrophy more prominent on upper palpebral conjunctiva.
Diffuse papillary hypertrophy more prominent on upper palpebral conjunctiva.
 Breakdown of conjunctival septae forming cobblestones (Figure 1.11A).
Breakdown of conjunctival septae forming cobblestones (Figure 1.11A).
 Bulbar conjunctival hyperemia.
Bulbar conjunctival hyperemia.
 Thickened gelatinous limbus with scattered opalescent mounds called Horner-Trantas dots (Figure 1-11C).
Thickened gelatinous limbus with scattered opalescent mounds called Horner-Trantas dots (Figure 1-11C).
 Fine punctate epithelial erosions: “flour dusting.”
Fine punctate epithelial erosions: “flour dusting.”
 Superior corneal pannus, may progress to 360 degrees.
Superior corneal pannus, may progress to 360 degrees.
 Shield ulcer (Figure 1.11B).
Shield ulcer (Figure 1.11B).
 Pseudogerontoxon: whitish partial stromal arcus.
Pseudogerontoxon: whitish partial stromal arcus.
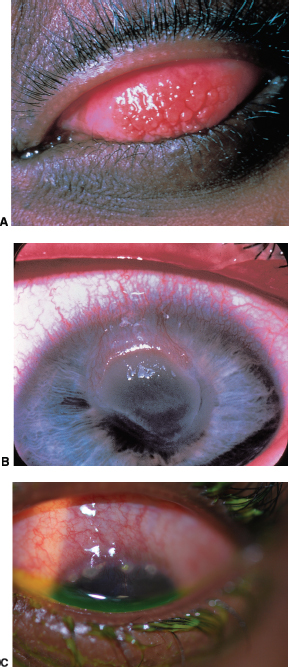
FIGURE 1.11. A: “Cobblestone” giant papillae in vernal conjunctivitis. B: Shield ulcer on the cornea in another patient with vernal conjunctivitis. C: Horner-Trantas dots in limbal vernal keratoconjunctivitis.
Systemic Findings
Atopic dermatitis (eczema).
Asthma.
Pathology
 Horner-Trantas dots: degenerated eosinophils in hypertrophied limbal tissue.
Horner-Trantas dots: degenerated eosinophils in hypertrophied limbal tissue.
 Infiltration of the conjunctival epithelium with eosinophils, plasma cells, lymphocytes, and monocytes.
Infiltration of the conjunctival epithelium with eosinophils, plasma cells, lymphocytes, and monocytes.
Disease Course
 Seasonal springtime exacerbations.
Seasonal springtime exacerbations.
 Often resolves by puberty.
Often resolves by puberty.
Treatment and Management
Antihistamines.
Mast cell stabilizers.
Oral topical and supratarsal steroids.
Topical cyclosporin A.
Climatotherapy: home air-conditioning or relocate to cooler climate.
Differential Diagnosis
Atopic keratoconjunctivitis.
Giant papillary conjunctivitis secondary to contact lenses.
Reference
Smolin G. Ocular allergy: The Third Annual Thygeson Lecture. Cornea 1998;17:253–256.
Infectious Conjunctivitis
Neonatal Conjunctivitis
Other name: ophthalmia neonatorum.
Summary
Neonatal conjunctivitis occurs within the first month of life. The most common causes include chemical irritation and infections with microorganisms including Neisseria gonorrhoeae (Figure 1.12), HSV, Chlamydia, and other organisms (Table 1.5).
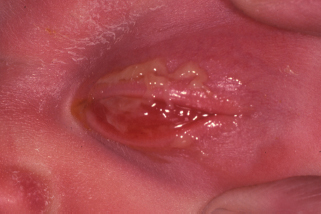
FIGURE 1.12. Gonococcal conjunctivitis in a neonate. (Courtesy of Mark Greenwald.)
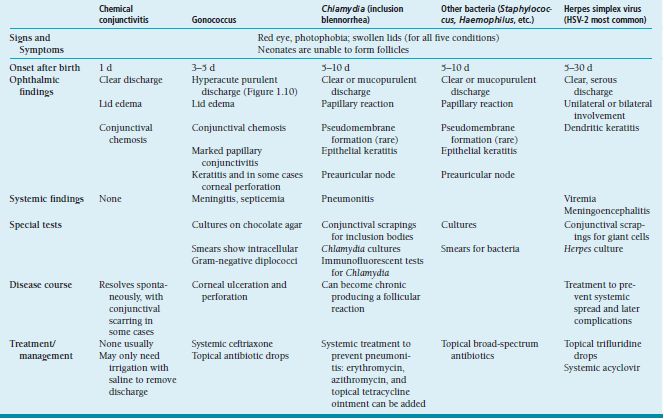
Etiology
 Chemical: postpartum instillation of drops or ointment (usually from the use of silver nitrate) used for prophylaxis against infection.
Chemical: postpartum instillation of drops or ointment (usually from the use of silver nitrate) used for prophylaxis against infection.
 Infectious: ocular contact with infected vaginal secretions.
Infectious: ocular contact with infected vaginal secretions.
 Be suspicious in mothers with premature rupture of membranes.
Be suspicious in mothers with premature rupture of membranes.
Demographics
Newborns.
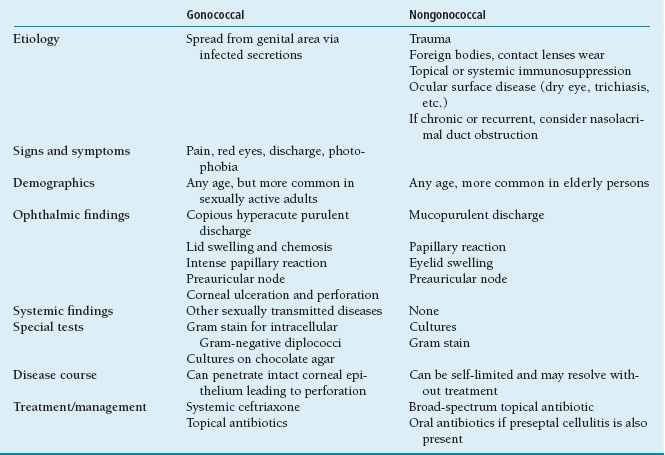
Bacterial Conjunctivitis
Summary
Conjunctivitis caused by bacteria is very common and in most cases is benign and self-limited. There is considerable overlap in the presenting findings from different bacteria. It is important to rule out possible infection by N. gonorrhoeae or N. meningitidis (Table 1.6). The most common etiologic organisms of bacterial conjunctivitis are Streptococcus pneumoniae, Staphylococcus aureus, and Haemophilus influenzae.
Viral Conjunctivitis
Summary
The most common cause of viral conjunctivitis is adenovirus (EKC); however, other viruses, including HSV, coxsackievirus, Enterovirus, and Newcastle disease viruses can affect the conjunctiva. Conjunctivitis from other viruses with conditions such as measles, influenza, and mumps is much less common (Table 1.7).
Chlamydia Conjunctivitis
Adult Inclusion Conjunctivitis
Summary
Adult inclusion conjunctivitis is a sexually transmitted conjunctivitis caused by Chlamydia. It should be considered in any case of chronic follicular conjunctivitis.
Etiology
 Spread by contact with infected genital secretions.
Spread by contact with infected genital secretions.
 Chlamydia trachomatis serotypes D to K.
Chlamydia trachomatis serotypes D to K.
Table 1.7 Viral conjunctivitis
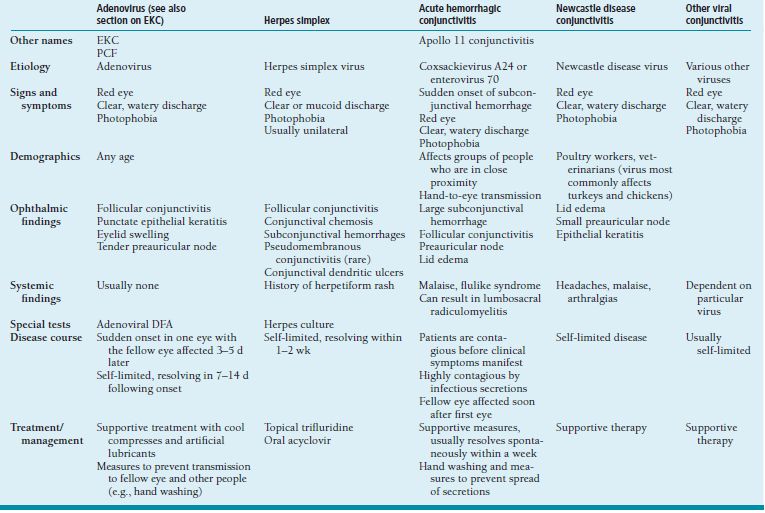
EKC, epidemic keratoconjunctivitis; PCF, pharyngoconjunctival fever; DFA, direct fluorescent antibody.
Signs and Symptoms
Acute onset of conjunctivitis.
Irritation.
Demographics
Young, sexually active teenagers and adults.
Ophthalmic Findings
Unilateral or bilateral conjunctivitis.
Tarsal follicles, often more prominent inferiorly.
Small, nontender preauricular node.
Punctate epithelial keratopathy (PEK).
Superficial pannus.
Chronic mucopurulent discharge.
Systemic Findings
 Chlamydial genital infection, may be asymptomatic.
Chlamydial genital infection, may be asymptomatic.
 Other sexually transmitted diseases.
Other sexually transmitted diseases.
Special Tests
 Giemsa stain for intracytoplasmic inclusions.
Giemsa stain for intracytoplasmic inclusions.
 Chlamydia organisms on direct fluorescent antibody (DFA) assay.
Chlamydia organisms on direct fluorescent antibody (DFA) assay.
 Cultures for Chlamydia organisms.
Cultures for Chlamydia organisms.
Disease Course
Chronic irritation and mild mucopurulent discharge.
Treatment and Management
Treatment of choice is systemic antibiotics:
1. Oral azithromycin.
2. Tetracycline.
3. Erythromycin.
4. Doxycycline.
Also treat sexual partners who may also be harboring chlamydial infection.
Differential Diagnosis
EKC.
Toxicity.
Molluscum contagiosum.
Psittacosis.
Trachoma
Summary
Trachoma, caused by C. trachomatis, is a major cause of blindness worldwide. Infection leads to conjunctival and corneal scarring, trichiasis, and dry eye.
Etiology
 Spread by contact with infected ocular secretions; can be spread by flies and other fomites.
Spread by contact with infected ocular secretions; can be spread by flies and other fomites.
 C. trachomatis serotypes A to C.
C. trachomatis serotypes A to C.
Demographics
 Found in hot, dry climates, often in underdeveloped countries.
Found in hot, dry climates, often in underdeveloped countries.
 Can occur in epidemics when many people live in close proximity.
Can occur in epidemics when many people live in close proximity.
Ophthalmic Findings
Early:
 Follicular conjunctivitis (Figure 1.13).
Follicular conjunctivitis (Figure 1.13).
 Mucopurulent discharge.
Mucopurulent discharge.
 Tender preauricular node.
Tender preauricular node.
 Punctate epithelial keratitis.
Punctate epithelial keratitis.
Late:
 Conjunctival scarring including Arlt line.
Conjunctival scarring including Arlt line.
 Entropion, trichiasis.
Entropion, trichiasis.
 Scarring of limbal follicles (Herbert pits).
Scarring of limbal follicles (Herbert pits).
 Superior superficial corneal neovascularization.
Superior superficial corneal neovascularization.
Systemic Findings
None.
Special Tests
Helpful only in acute infection: cultures, DFA.
Disease Course
Resolution of early infection with scarring occurring years to decades later.
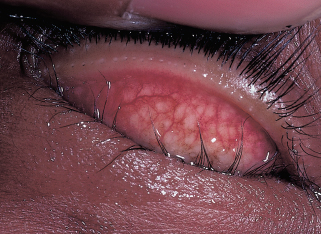
FIGURE 1.13. Follicular conjunctivitis in a 5-year-old child in a trachoma endemic area.
Treatment and Management
Early treatment:
 Oral azithromycin.
Oral azithromycin.
 Topical tetracycline, azithromycin, or erythromycin.
Topical tetracycline, azithromycin, or erythromycin.
Late treatment:
 Management of complications such as dry eye and bacterial superinfection.
Management of complications such as dry eye and bacterial superinfection.
 Surgery for trichiasis or entropion.
Surgery for trichiasis or entropion.
Cicatricial Conjunctivitis
Differential Diagnosis
 Any severe infection producing a membrane or pseudomembrane:
Any severe infection producing a membrane or pseudomembrane:
Bacterial (Neisseria, beta-hemolytic Streptococcus, Corynebacterium, Haemophilus organisms).
Viral (EKC, HSV).
Chlamydial.
 Systemic medications:
Systemic medications:
Practolol.
D-penicillinamine.
 Topical medications: particularly glaucoma medications such as epinephrine and pilocarpine.
Topical medications: particularly glaucoma medications such as epinephrine and pilocarpine.
 Dermatobullous diseases:
Dermatobullous diseases:
Toxic epidermal necrolysis.
Dermatitis herpetifomis.
Pemphigus vulgaris.
Porphyria cutanea tarda.
 Autoimmune:
Autoimmune:
Stevens-Johnson syndrome.
Ocular cicatricial pemphigoid.
Systemic lupus erythematosus.
Sarcoidosis.
Atopic blepharoconjunctivitis.
Progressive systemic sclerosis.
Sjön syndrome.
 Trauma:
Trauma:
Chemical and thermal burns.
Conjunctival surgery.
Conjunctival injury.
Radiation exposure.
Reference
Holsclaw DS. Ocular cicatricial pemphigoid. Int Ophthalmol Clin 1998;38:95.
Ocular Cicatricial Pemphigoid
Summary
OCP is an autoimmune disease of unknown etiology affecting the skin and mucosa. The mainstay of treatment is immunosuppression, as well as prompt diagnosis and treatment of the many ocular complications of the disease (Figure 1.14).
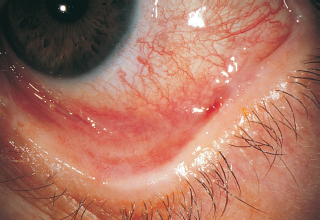
FIGURE 1.14. Ocular pemphigoid. Severe conjunctival foreshortening and symblepharon in a patient with moderately advanced ocular cicatricial pemphigoid. (From Tasman W, Jaeger E. The Wills Eye Hospital atlas of clinical ophthalmology, 2nd ed. Philadelphia, PA: Lippincott Williams &Wilkins, 2001.)
Etiology
 Autoimmune.
Autoimmune.
 Circulating antibodies and complement bind to the basement membrane of mucosal tissue at sites throughout the body. The initial stimulus for the formation of these autoreactive antibodies is not understood.
Circulating antibodies and complement bind to the basement membrane of mucosal tissue at sites throughout the body. The initial stimulus for the formation of these autoreactive antibodies is not understood.
Signs and Symptoms
 Dry eye symptoms such as foreign body sensation.
Dry eye symptoms such as foreign body sensation.
 Chronic red eye.
Chronic red eye.
Demographics
 Mean age of presentation is in the seventh decade of life.
Mean age of presentation is in the seventh decade of life.
 The disease may rarely present in the pediatric population.
The disease may rarely present in the pediatric population.
 Women are affected twice as frequently as men.
Women are affected twice as frequently as men.
Ophthalmic Findings
Chronic bilateral, papillary, cicatricial conjunctivitis with symblepharon formation (Figure 1.15A).
Forniceal foreshortening.
Diminished tear production.
Trichiasis.
Conjunctival keratinization.
Corneal ulcers, neovascularization, and scarring.
Systemic Findings
 Any mucosal surface may be affected in cicatrical pemphigoid.
Any mucosal surface may be affected in cicatrical pemphigoid.
 Oral mucosa is involved in 65% of patients with OCP, such as desquamative gingivitis (Figure 1.15B).
Oral mucosa is involved in 65% of patients with OCP, such as desquamative gingivitis (Figure 1.15B).
 Involvement of the larynx and esophagus may lead to life-threatening complications; it is important to elicit a history of changes in speech or swallowing in patients with OCP.
Involvement of the larynx and esophagus may lead to life-threatening complications; it is important to elicit a history of changes in speech or swallowing in patients with OCP.
 Vesiculobullous skin lesions that resolve without scarring, or erythematous plaques, which do scar.
Vesiculobullous skin lesions that resolve without scarring, or erythematous plaques, which do scar.
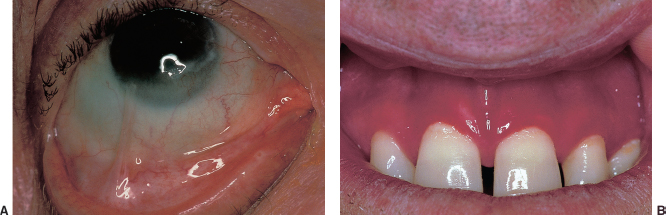
FIGURE 1.15. A: Symblephera in a patient with severe ocular cicatricial pemphigoid. B: Oral manifestations of pemphigoid include oral ulceration and gingivitis.
Special Tests
Biopsy of the oral or conjunctival mucosa with direct immunofluorescence to detect the presence of linear deposition of immunoglobulin and complement on the basement membrane. This test is not available in all pathology departments. Specimens must be processed when fresh and must not be placed in formalin. Therefore, before undertaking a biopsy, arrangements should be made for the proper handling and interpretation of the specimen.
Pathology
Linear deposition of immunoglobin, complement, and fibrinogen on the basement membrane of the conjunctival mucosa.
Disease Course
 Wide range in severity from mild to vision threatening.
Wide range in severity from mild to vision threatening.
 In severe cases, the combination of diminished tear production and trichiasis leads to recurrent corneal ulcers and corneal scarring that may be blinding.
In severe cases, the combination of diminished tear production and trichiasis leads to recurrent corneal ulcers and corneal scarring that may be blinding.
Treatment and Medications
 After a diagnosis of active OCP has been confirmed by tissue biopsy, systemic immunosuppressive therapy should be initiated in most cases.
After a diagnosis of active OCP has been confirmed by tissue biopsy, systemic immunosuppressive therapy should be initiated in most cases.
 Immunosuppressive therapy with cyclophosphamide is the mainstay, but this is an evolving field of study.
Immunosuppressive therapy with cyclophosphamide is the mainstay, but this is an evolving field of study.
 Methotrexate, azathioprine, mycophenolate, and other immunosupressive agents have been used for patients who cannot tolerate cyclophosphamide.
Methotrexate, azathioprine, mycophenolate, and other immunosupressive agents have been used for patients who cannot tolerate cyclophosphamide.
 Emerging therapies such as rituximab and intravenous immunoglobulin have been advocated by some.
Emerging therapies such as rituximab and intravenous immunoglobulin have been advocated by some.
 Often, patients are treated with prednisone while cyclophosphamide therapy is being initiated.
Often, patients are treated with prednisone while cyclophosphamide therapy is being initiated.
 The prednisone is withdrawn once the cyclophosphamide has reached a therapeutic level.
The prednisone is withdrawn once the cyclophosphamide has reached a therapeutic level.
 Immunosuppressive treatment is usually continued for 9 to 12 months.
Immunosuppressive treatment is usually continued for 9 to 12 months.
 Consultation with an internist for assistance in the management of these immunosuppressive medications and the development of extraocular manifestations of the disease is important.
Consultation with an internist for assistance in the management of these immunosuppressive medications and the development of extraocular manifestations of the disease is important.
 Formal OCP staging systems and serial photographs are useful to monitor progression of disease.
Formal OCP staging systems and serial photographs are useful to monitor progression of disease.
 Definitive surgical treatment of any trichiasis or other procedures should be undertaken when the eye is in remission during immunosuppressive therapy.
Definitive surgical treatment of any trichiasis or other procedures should be undertaken when the eye is in remission during immunosuppressive therapy.
 Specific ocular complications of OCP such as dry eye, keratinization, and corneal ulcers should be treated as they arise.
Specific ocular complications of OCP such as dry eye, keratinization, and corneal ulcers should be treated as they arise.
Differential Diagnosis
Conjunctival scarring and trichiasis may be seen in association with many conditions:
 Bacterial and viral conjunctivitis.
Bacterial and viral conjunctivitis.
 Chlamydial infection.
Chlamydial infection.
 Long-term use of glaucoma medications.
Long-term use of glaucoma medications.
 Atopic keratoconjunctivitis.
Atopic keratoconjunctivitis.
 Chemical or thermal burns.
Chemical or thermal burns.
 Radiation exposure.
Radiation exposure.
 Trauma.
Trauma.
 Sarcoidosis.
Sarcoidosis.
 SJS.
SJS.
 Other dermatobullous disorders.
Other dermatobullous disorders.
References
Bernauer W, Elder MJ, Leonard JN, et al. The value of biopsies in the evaluation of chronic progressive conjunctival cicatrisation. Graefes Arch Clin Exp Ophthalmol 1994;32:533–537.
Elder MJ, Lightman S, Dart JKG. Role of cyclophosphamide and high dose steroid in ocular cicatricial pemphigoid. Br J Ophthalmol 1995;79:264–266.
Foster CS. Cicatricial pemphigoid. Trans Am Ophthalmol Soc 1986;84:527–663.
Foster CS, Chang PY, Ahmed AR. Combination of Rituximab and intravenous immunoglobulin for recalcitrant ocular cicatricial pemphigoid: a preliminary report. Ophthalmology 2010;117(5):861–869.
Holsclaw DS. Ocular cicatricial pemphigoid. Int Ophthalmol Clin 1998;38:89–106.
Saw VP, Dart JK, Rauz S, et al. Immunosuppressive therapy for ocular mucous membrane pemphigoid strategies and outcomes. Ophthalmology 2008;115(2):253–261.e1.
Tauber J, Sainz de la Maza M, Foster CS. Systemic chemotherapy for ocular cicatricial pemphigoid. Cornea 1991;10:185–195.
Stevens-Johnson Syndrome
Other name: erythema multiforme major.
Summary
Acute mucocutaneous eruption usually associated with medication or infection, with a prodrome of malaise and fever, followed by erythematous skin lesions involving 10% to 20% of body surface area and two mucosal sites.
Etiology
Unknown, but most of the cases are drug related or postinfectious (Table 1.8).
Signs and Symptoms
 Malaise and fever.
Malaise and fever.
 Cutaneous and mucosal eruptions.
Cutaneous and mucosal eruptions.
 Red eyes.
Red eyes.
 Photophobia.
Photophobia.
Demographics
 Male-to-female predominance of 3 to 1.
Male-to-female predominance of 3 to 1.
 Usually occurring in people between 10 and 30 years of age.
Usually occurring in people between 10 and 30 years of age.
 SJS may be more common among people with AIDS.
SJS may be more common among people with AIDS.

Ophthalmic Findings
Acute stage of the disease:
 Desquamative bullous rash of the skin of the eyelids.
Desquamative bullous rash of the skin of the eyelids.
 Mucopurulent conjunctivitis.
Mucopurulent conjunctivitis.
 Episcleritis.
Episcleritis.
 Severe papillary conjunctivitis.
Severe papillary conjunctivitis.
 Ulcerative conjunctivitis with pseudomembranes.
Ulcerative conjunctivitis with pseudomembranes.
Chronic ocular complications:
 Cicatricial conjunctivitis.
Cicatricial conjunctivitis.
 Trichiasis.
Trichiasis.
 Entropion.
Entropion.
 Symblepharon.
Symblepharon.
 Dry eye.
Dry eye.
 Keratinization.
Keratinization.
 Corneal infections and scarring.
Corneal infections and scarring.
Progressive conjunctival scarring may occur even after the acute systemic disease has resolved.
Systemic Findings
 Skin lesions: erythematous macules or papules smaller than 3 mm that classically develop a target appearance. As the disease progresses, these lesions may coalesce. Despite their dramatic appearance, the cutaneous manifestations of SJS often resolve without sequelae.
Skin lesions: erythematous macules or papules smaller than 3 mm that classically develop a target appearance. As the disease progresses, these lesions may coalesce. Despite their dramatic appearance, the cutaneous manifestations of SJS often resolve without sequelae.
 Other mucosal surfaces in the body, particularly the oral mucosa, may become involved.
Other mucosal surfaces in the body, particularly the oral mucosa, may become involved.
 SJS may become a life-threatening condition as a result of superinfection, electrolyte imbalance, or severe volume depletion.
SJS may become a life-threatening condition as a result of superinfection, electrolyte imbalance, or severe volume depletion.
Special Tests
Usually none. A biopsy can be performed in cases that are not clinically obvious.
Pathology
Dermal and epidermal lymphocytic and histocytic infiltrate. Eosinophils may be seen in patients with drug-induced SJS.
Treatment and Management
 The systemic manifestation of SJS often makes the patient’s condition medically unstable, and admission to a medical ward or burn unit may be required.
The systemic manifestation of SJS often makes the patient’s condition medically unstable, and admission to a medical ward or burn unit may be required.
 Patients with ulcerative conjunctivitis and/or epithelial corneal defects should receive prophylactic topical antibiotics and should be monitored for infection.
Patients with ulcerative conjunctivitis and/or epithelial corneal defects should receive prophylactic topical antibiotics and should be monitored for infection.
 Bandage contact lenses, plastic wrap, symblepharon rings, amniotic membrane, and daily lysis of symblepharon have all been advocated for the prevention of symblepharon.
Bandage contact lenses, plastic wrap, symblepharon rings, amniotic membrane, and daily lysis of symblepharon have all been advocated for the prevention of symblepharon.
 Topical and systemic steroids are often used during the acute phase of the disease, but their use is controversial.
Topical and systemic steroids are often used during the acute phase of the disease, but their use is controversial.
 Corneal transplantation has a poor prognosis. Keratoprosthesis has been used with some success.
Corneal transplantation has a poor prognosis. Keratoprosthesis has been used with some success.
 Amniotic membrane transplantation and limbal stem cell transplantation have been used.
Amniotic membrane transplantation and limbal stem cell transplantation have been used.
 The ocular chronic complications of SJS are often the most troublesome sequelae of the disease. Trichiasis, entropion, symblepharon, dry eye, keratinization, and corneal infection and opacification must all be managed as they arise.
The ocular chronic complications of SJS are often the most troublesome sequelae of the disease. Trichiasis, entropion, symblepharon, dry eye, keratinization, and corneal infection and opacification must all be managed as they arise.
References
Foster CS, Fong LP, Azar D, et al. Episodic conjunctival inflammation after Stevens-Johnson syndrome. Ophthalmology 1988;95:453–462.
Holland EG, Palmon FE, Webster GF. Erythema multiforme, Stevens-Johnson syndrome and toxic epidermal necrolysis. In: Mannis MJ, Macsai MS, Huntley AC, eds. Eye and skin disease. New York, NY: Lippincott-Raven Publishers, 1996:273–284.
Mondino BJ. Cicatricial pemphigoid and erythema multiforme. Ophthalmology 1990;97:939–952.
Sayegh RR, Ang LP, Foster CS, et al. The Boston keratoprosthesis in Stevens-Johnson syndrome. Am J Ophthalmol 2008; 145(3):438–444.
Graft Versus Host Disease
Summary
Graft versus host disease (GVHD) is a severe complication following bone marrow transplantation in which transplanted immunocompetent cells attack the host tissues. Ocular involvement during the acute phase can range from conjunctival erythema to frank epithelial sloughing. Late complications include scarring and dry eye.
Etiology
Transplanted immune cells from the donor reacting against the immunoincompetent host following bone marrow transplantation.
Signs and Symptoms
Acute:
Red eyes.
Photophobia.
Serous or bloody tears.
Membrane formation.
Chronic:
Dry eye.
Foreign body sensation.
Chronic red eye.
Superior limbic keratoconjunctivitis.
Demographics
 Patients after bone marrow transplantation.
Patients after bone marrow transplantation.
 Acute phase (within 100 days), chronic phase (after 100 days).
Acute phase (within 100 days), chronic phase (after 100 days).
Ophthalmic Findings
Acute:
 Conjunctival hyperemia and papillary reaction.
Conjunctival hyperemia and papillary reaction.
 Chemosis and serosanguineous exudation.
Chemosis and serosanguineous exudation.
 Membrane or pseudomembrane formation.
Membrane or pseudomembrane formation.
 Epithelial sloughing.
Epithelial sloughing.
 Heals with cicatrization, symblephera.
Heals with cicatrization, symblephera.
Chronic:
 Cicatricial conjunctival changes similar to OCP.
Cicatricial conjunctival changes similar to OCP.
 Dry eye.
Dry eye.
 Superior Limbic Keratoconjunctivitis.
Superior Limbic Keratoconjunctivitis.
Systemic Findings
Skin: maculopapular rash with bulla formation.
Liver: elevated liver enzyme and bilirubin values.
Gut: diarrhea.
Pathology
 Subepithelial lymphocytic infiltrate with bulla formation.
Subepithelial lymphocytic infiltrate with bulla formation.
 Lymphocytic infiltration of the lacrimal gland.
Lymphocytic infiltration of the lacrimal gland.
Disease Course
 Progressive conjunctival and corneal scarring with symblephera formation, entropion, and trichiasis.
Progressive conjunctival and corneal scarring with symblephera formation, entropion, and trichiasis.
 Dry eye with lacrimal gland involvement and loss of goblet cells.
Dry eye with lacrimal gland involvement and loss of goblet cells.
Treatment and Management
 High-dose systemic steroids.
High-dose systemic steroids.
 Local treatment for superinfection, dry eyes.
Local treatment for superinfection, dry eyes.
Differential Diagnosis
OCP.
Reference
Jabs DA, Wingard JR, Green WR, et al. The eye in bone marrow transplantation, III: conjunctival graft-versus-host disease. Arch Ophthalmol 1989;1007:1343–1348.
Chemical Burns
See p. 74.
Miscellaneous Causes of Conjunctivitis
Floppy Eyelid Syndrome
Summary
Floppy eyelid syndrome occurs primarily in overweight individuals and is characterized by softening of the upper tarsus and laxity allowing easy eversion, especially while sleeping, causing chronic irritation and inflammation.
Etiology
 Extreme laxity of the upper eyelid with spontaneous eversion.
Extreme laxity of the upper eyelid with spontaneous eversion.
 Direct contact of the everted lids with bedsheets leads to chronic irritation.
Direct contact of the everted lids with bedsheets leads to chronic irritation.
Signs and Symptoms
Redness.
Irritation.
Discharge.
Demographics
Overweight individuals.
No sex predilection.
Ophthalmic Findings
 Upper lid laxity and floppy tarsus (Figure 1.16).
Upper lid laxity and floppy tarsus (Figure 1.16).
 Confluent small to large papillae on the upper palpebral conjunctiva.
Confluent small to large papillae on the upper palpebral conjunctiva.
 Mucous discharge.
Mucous discharge.
 Punctate corneal epitheliopathy.
Punctate corneal epitheliopathy.
 Superficial corneal vascularization.
Superficial corneal vascularization.
 May be unilateral if patient sleeps on one side.
May be unilateral if patient sleeps on one side.
Systemic Findings
Obesity.
Sleep apnea syndrome.
Special Tests
None.
Disease Course
 Chronic and persistent before intervention.
Chronic and persistent before intervention.
 May be easily confused with other conditions such as medicamentosa and allergy.
May be easily confused with other conditions such as medicamentosa and allergy.
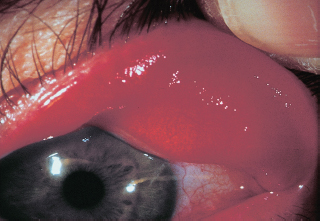
FIGURE 1.16. Spontaneous eyelid eversion with gentle elevation of the eyelid. Notice the tarsal kink with loss of its normal fibrous rigidity.
Treatment and Management
 Cover affected eye with shield at night.
Cover affected eye with shield at night.
 Patch eye at night.
Patch eye at night.
 Surgical tightening procedure may be necessary.
Surgical tightening procedure may be necessary.
Medications
Lubricants.
Differential Diagnosis
Vernal conjunctivitis.
Giant papillary conjunctivitis.
Atopic conjunctivitis.
Bacterial conjunctivitis.
Toxic conjunctivitis.
References
Culbertson W, Ostler W. Floppy eyelid syndrome. Am J Ophthalmol 1981;92:568–575.
McNab AA. Floppy eyelid syndrome and obstructive sleep apnea. Ophthal Plast Reconstr Surg 1997;13:98–114.
Giant Papillary Conjunctivitis
Summary
Giant papillary conjunctivitis is a descriptive term for a conjunctivitis caused by mechanical irritation, most frequently by contact lens use. The hallmark is the presence of giant papillae on the superior tarsal conjunctiva.
Etiology
Chronic mechanical trauma or irritation of the conjunctiva by contact lenses, exposed sutures, or prostheses.
Signs and Symptoms
 Itchy eyes.
Itchy eyes.
 Mucoid discharge, worse when wearing lenses.
Mucoid discharge, worse when wearing lenses.
 Decreased comfort and wearing time of lenses.
Decreased comfort and wearing time of lenses.
Demographics
Contact lens wearers, more common in patients with poor lens hygiene or overnight wear.
Ophthalmic Findings
 Giant papillae on superior tarsal conjunctiva with apical scarring.
Giant papillae on superior tarsal conjunctiva with apical scarring.
 Mucoid discharge.
Mucoid discharge.
Systemic Findings
None.
Disease Course
May resolve spontaneously with removal of irritant but may leave residual scars.
Treatment and Management
 Discontinuation of contact lens wear.
Discontinuation of contact lens wear.
 Change of lenses to disposable soft contact lenses, low-water-content lenses, or gas-permeable hard contact lenses.
Change of lenses to disposable soft contact lenses, low-water-content lenses, or gas-permeable hard contact lenses.
 Topical mast cell stabilizers.
Topical mast cell stabilizers.
 Increased enzyme tablet use.
Increased enzyme tablet use.
 Topical steroids, if condition is severe.
Topical steroids, if condition is severe.
Differential Diagnosis
Vernal conjunctivitis.
Dry Eyes
Keratoconjunctivitis Sicca
Other names: Sjön syndrome (KCS with dry mouth).
Summary
Dry eyes is an extremely common ophthalmic condition caused by decreased tear production, increased tear evaporation, or instability of the tear film. Many associated conditions can result in dry eyes, making diagnosis and treatment more complicated.
Etiology
Three major causes:
1. Decreased tear production:
Sjön syndrome.
Non-Sjön syndrome.
Infiltration of the lacrimal gland such as with sarcoidosis.
Decreased corneal sensation.
Use of certain medications (anticholinergics, antihistamines).
2. Evaporative dry eye:
Meibomitis.
Goblet cell loss (SJS, cicatricial pemphigoid).
Exogenous irritants.
Abnormal lid anatomy.
Thyroid eye disease.
Cosmetic lid surgery.
Lagophthalmos.
Low-humidity environments (airplanes, air-conditioned rooms, desert climate).
Signs and Symptoms
Sandy, gritty sensation.
Constant foreign body sensation.
Redness.
Blurred vision.
Worse symptoms with wind and dry climates.
Demographics
 More common in elderly individuals and in women.
More common in elderly individuals and in women.
 Patients with thyroid eye disease or systemic autoimmune diseases may also be predisposed.
Patients with thyroid eye disease or systemic autoimmune diseases may also be predisposed.
Ophthalmic Findings
 Increased interpalpebral fissure height.
Increased interpalpebral fissure height.
 Lagophthalmos or incomplete blink.
Lagophthalmos or incomplete blink.
 Decreased tear lake.
Decreased tear lake.
 Lissamine green or rose bengal staining of conjunctiva (Figure 1.17); fluorescein staining of cornea.
Lissamine green or rose bengal staining of conjunctiva (Figure 1.17); fluorescein staining of cornea.
 Keratinization of the cornea or conjunctiva.
Keratinization of the cornea or conjunctiva.
 Superficial corneal neovascularization.
Superficial corneal neovascularization.
 Blepharitis or meibomitis.
Blepharitis or meibomitis.
 Excessive mucus formation.
Excessive mucus formation.
 Corneal filaments.
Corneal filaments.
 Malposition of the puncta.
Malposition of the puncta.
Systemic Findings
Sjön syndrome: systemic sicca condition with dry mouth, dry skin, trouble swallowing, often associated with autoimmune diseases such as rheumatoid arthritis, SLE.
Special Tests
Schirmer testing: with or without topical anesthesia. Standardized strips of filter paper absorb tears. Consistent measurements of less than 5 mm of wetting at 5 minutes may be indicative of dry eyes.
Tear break-up time (TBUT): Instill fluorescein into the tear film. Observe the surface of the tear film for areas of disruption. Normal TBUT is generally greater than 10 seconds.
Treatment and Management
 Treat underlying cause (e.g., blepharitis, lid malposition, meibomitis).
Treat underlying cause (e.g., blepharitis, lid malposition, meibomitis).
 Aqueous tear replacements.
Aqueous tear replacements.
 Punctal occlusion or tarsorrhaphy in severe cases.
Punctal occlusion or tarsorrhaphy in severe cases.
 Decrease evaporative loss with increased humidity, moist chambers, or ointments.
Decrease evaporative loss with increased humidity, moist chambers, or ointments.
 Topical cyclosporine.
Topical cyclosporine.
 Topical steroids.
Topical steroids.
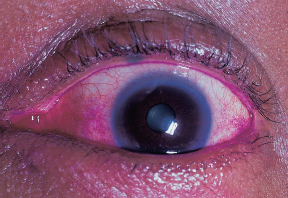
FIGURE 1.17. Interpalpebral rose bengal staining in a patient with KCS.
Differential Diagnosis
 Meibomitis.
Meibomitis.
 Exposure keratopathy.
Exposure keratopathy.
 Toxic keratopathy: Patients may be sensitive to preservatives in many over-the-counter eyedrops.
Toxic keratopathy: Patients may be sensitive to preservatives in many over-the-counter eyedrops.
Vitamin A Deficiency
Summary
Without vitamin A, mucous membranes undergo squamous metaplasia with loss of goblet cells and subsequent xerosis. This condition is more common in developing countries where the diet may be nutritionally deficient.
Etiology
Deficiency of vitamin A leads to abnormal development of mucous membranes and metaplasia from columnar epithelium to stratified squamous epithelium.
Signs and Symptoms
Sandy, gritty irritation.
Foreign body sensation.
Demographics
 Nutritional deficiency:
Nutritional deficiency:
Malnourished children in developing countries.
Fad diets and alcoholics.
 Fat malabsorption syndromes (e.g., cystic fibrosis): Vitamin A is a fat-soluble vitamin.
Fat malabsorption syndromes (e.g., cystic fibrosis): Vitamin A is a fat-soluble vitamin.
Ophthalmic Findings
 Conjunctival metaplasia to stratified squamous epithelium with loss of goblet cells.
Conjunctival metaplasia to stratified squamous epithelium with loss of goblet cells.
 White keratinized conjunctival plaque (Bitot spot).
White keratinized conjunctival plaque (Bitot spot).
 Corneal drying and diffuse, confluent punctate keratopathy.
Corneal drying and diffuse, confluent punctate keratopathy.
 Corneal ulceration and keratomalacia.
Corneal ulceration and keratomalacia.
 Bacterial superinfection.
Bacterial superinfection.
 Nyctalopia (night blindness) (vitamin A is necessary for the production of rhodopsin and phototransduction).
Nyctalopia (night blindness) (vitamin A is necessary for the production of rhodopsin and phototransduction).
Systemic Findings
Metaplasia of mucous membranes of the lung and gut.
Systemic vitamin A deficiency has a 50% untreated mortality rate.
Disease Course
High incidence of bacterial superinfection and ulceration.
Treatment and Management
 Oral vitamin A supplementation.
Oral vitamin A supplementation.
 Topical vitamin A ointment (may be of limited benefit).
Topical vitamin A ointment (may be of limited benefit).
CONGENITAL CORNEAL ABNORMALITIES
Anterior Segment Dysgenesis
Summary
A group of congenital abnormalities of the anterior segment involving developmental arrest or incomplete migration of mesodermal or neural crest tissues (Table 1.9).
Signs and Symptoms
External examination in the newborn period may reveal abnormalities, prompting referral. For milder asymptomatic abnormalities, diagnosis may be made at the time of a routine examination. Visual deficits are variable.

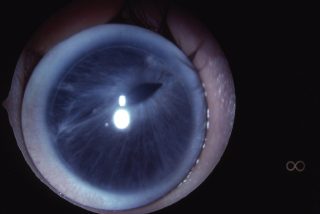
FIGURE 1.18. Anterior segment dysgenesis. (Courtesy of Mark Greenwald.)
Demographics
None specific.
Ophthalmic Findings and Systemic Findings
See Figure 1.18.
Posterior embryotoxon: prominent anteriorly displaced Schwalbe line (Figure 1.19A) visible on slit-lamp examination as a white arc that is central to the limbus. May be seen in up to 30% of normal individuals; causes no deficits. It is usually dominantly inherited.
Axenfeld-Rieger syndrome: Previous nomenclature has been compressed to include overlapping entities that are characterized by having posterior embryotoxon, iris strands (Figure 1.19B), iris and pupillary abnormalities (Figure 1.19C), and glaucoma in 50%. Often associated with dental, cranial, facial, or skeletal abnormalities. Axenfeld-Reiger syndrome is most often autosomal dominant, but it may be sporadic.
Peters anomaly: central corneal opacity, usually with strands of adherent iris. The lens may have an anterior polar cataract or keratolenticular adhesion (Figure 1.19D). Glaucoma is common. Associated ocular abnormalities include microphthalmos, persistent hyperplastic primary vitreous, and retinal dysplasia. In 60% of cases, systemic abnormalities may be found: mental retardation, urogenital, craniofacial, and skeletal abnormalities. Sixty percent of cases are bilateral.
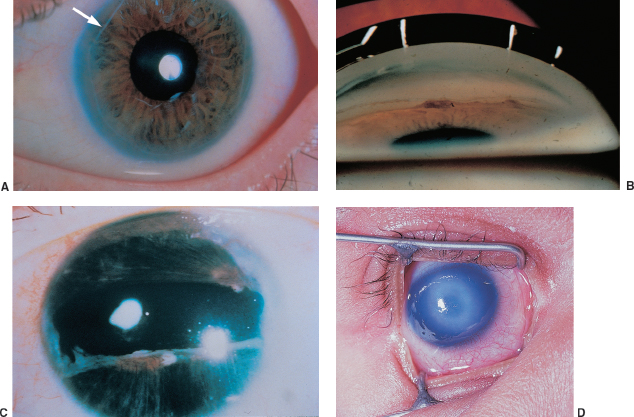
FIGURE 1.19. A: Posterior embryotoxon (arrow) is a congenital anterior displacement of Schwalbe line. B: Broad peripheral iridocorneal adhesions to Schwalbe line is found in Axenfeld anomaly. C: Further along the spectrum of anterior segment dysgenesis is Rieger anomaly with iris abnormalities. D: Central corneal opacity corresponding to an area of absent Descemet membrane. In many cases, the iris is adherent to the edge of this defect.
Special Tests
Gonioscopy should be included in the ocular examination if one of these abnormalities is suspected. Anterior segment ultrasound may be helpful to determine the lens position in Peters anomaly if surgery is considered.
Pathology
Peters anomaly is remarkable for a focal absence of Descemet membrane and endothelium in the area of central opacity.
Disease Course
Nonprogressive.
Treatment and Management
 Secondary complications such as glaucoma and amblyopia require ongoing care and follow-up.
Secondary complications such as glaucoma and amblyopia require ongoing care and follow-up.
 Peters anomaly may require penetrating keratoplasty (PKP).
Peters anomaly may require penetrating keratoplasty (PKP).
Differential Diagnosis
Other causes of congenital corneal opacities should be considered in the differential diagnosis of Peters anomaly.
Reference
Waring GO III, Rodriques MM, Labison PR. Anterior chamber cleavage syndromes: a stepladder classification. Surv Ophthalmol 1975;20:3–27.
Aniridia
Summary
Aniridia is a bilateral developmental abnormality of the iris. A variably sized stump of iris is present in the periphery, which, if small, is obscured by overlying sclera, giving the appearance of no iris.
Etiology
 Familial, autosomal dominant in 85% of cases.
Familial, autosomal dominant in 85% of cases.
 Fifteen percent are sporadic cases, attributable to abnormality of the short arm of chromosome 11 (Miller syndrome).
Fifteen percent are sporadic cases, attributable to abnormality of the short arm of chromosome 11 (Miller syndrome).
 Two percent are familial, autosomal recessive: termed Gillespie syndrome.
Two percent are familial, autosomal recessive: termed Gillespie syndrome.
Signs and Symptoms
Photophobia, pendular nystagmus, poor vision.
Demographics
None specific.
FIGURE 1.20. Retroillumination of the cornea of an aniridic patient with a rudimentary iris and superficial corneal vascularization.
Ophthalmic Findings
Variably hypoplastic iris.
Superficial corneal pannus due to limbal stem cell deficiency (Figure 1.20).
Foveal hypoplasia.
Amblyopia.
Strabismus.
High frequency of glaucoma, superficial corneal vascularization, and opacification.
Lens abnormalities and optic nerve hypoplasia also seen.
Systemic Findings
Miller syndrome: Wilms tumor of the kidney, Aniridia, Genitourinary abnormalities, mental Retardation. (WAGR syndrome)
Gillespie Syndrome: structural abnormalities of cerebellum; do not develop Wilms tumor.
Special Tests
Family history to determine sporadic cases, which then require workup for Wilms tumor (intravenous pyelogram [IVP] or ultrasound).
Pathology
Hypoplasia of iris.
Disease Course
Stable.
Treatment and Management
Strabismus, amblyopia, and glaucoma require usual care.
Medications
None specific.
Follow-Up
Require indefinite follow-up for corneal changes, lens abnormalities, and glaucoma.
Reference
Nelson LB, Spaeth GC, Nowinski TS, et al. Aniridia: a review. Surv Ophthalmol 1984;28:621–642.
Iridocorneal Endothelial Syndromes
Summary
A group of nonfamilial unilateral syndromes marked by corneal and iris abnormalities with a high association with glaucoma.
Etiology
Corneal endotheliopathy in which endothelial cells can proliferate and migrate, causing endothelization of the iris.
Signs and Symptoms
None in early disease; decreased vision in Chandler syndrome as corneal edema develops; iris abnormalities may be noted as a cosmetic abnormality.
Demographics
More common in women, findings present from early adulthood.
Ophthalmic Findings
Differentiated into three individual syndromes (Table 1.10).
Systemic Findings
None.
Special Tests
Specular microscopy provides useful information regarding the status of the endothelium.
Pathology
Abnormality of cells lining the cornea, trabecular meshwork, and anterior iris surface, which elaborate excessive basement membrane material and may contract, causing iris and pupil abnormalities.
Disease Course
Slowly progressive. Glaucoma control is problematic because of endothelialization and descemetization of the angle and filtering blebs.
Treatment and Management
Glaucoma management. Conservative management of corneal edema with sodium chloride drops and ointments is recommended. Can recur after penetrating or endothelial transplantation.
Medications
As required for glaucoma and corneal edema.
Follow-Up
Regular examination is indicated to monitor for glaucoma and corneal edema.
Differential Diagnosis
Findings similar to those of the iridocorneal endothelial (ICE) syndromes may be seen bilaterally in posterior polymorphous membranous dystrophy and Fuchs dystrophy, and in Axenfeld and Rieger syndromes. Iris melanoma should be considered in the differential of true unilateral findings but is notable for its lack of other iris abnormalities that are seen in the ICE syndromes.
Reference
Shields MB. Progressive essential iris atrophy, Chandler syndrome and the iris nevus syndrome: a spectrum of disease. Surv Ophthalmol 1979;24:3–20.
Birth Trauma
Summary
Unilateral congenital clouding of the cornea attributable to injury at birth.

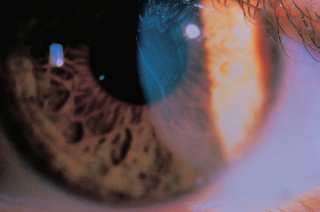
FIGURE 1.21. Vertically oriented Haab striae in a child with congenital glaucoma corresponding to ruptures of Descemet membrane secondary to the elevated pressures.
Etiology
Usually attributable to forceps delivery.
Signs and Symptoms
Cloudy cornea at birth, decreased vision as a result of high astigmatism later in life.
Demographics
None specific.
Ophthalmic Findings
 Acutely, corneal edema.
Acutely, corneal edema.
 Later, vertical or obliquely oriented breaks of Descemet membrane are seen, marked by scrolls or ridges of Descemet membrane (Figure 1.21).
Later, vertical or obliquely oriented breaks of Descemet membrane are seen, marked by scrolls or ridges of Descemet membrane (Figure 1.21).
 High degree of astigmatism and late endothelial decompensation are also seen.
High degree of astigmatism and late endothelial decompensation are also seen.
Systemic Findings
None.
Special Tests
None.
Pathology
Breaks, scrolls, ridges, and duplication of Descemet membrane may be seen.
Disease Course
See findings above.
Treatment and Management
Correction of astigmatism and amblyopia management.
Medications
Generally not required. Sodium chloride drops and ointment may speed short-term resolution of corneal edema.
Follow-Up
As required for amblyopia if present. Yearly examinations, at minimum, during childhood to monitor vision and refraction.
Differential Diagnosis
Congenital glaucoma may also present with breaks in Descemet membrane and corneal edema; breaks are usually peripheral and arcuate or paracentral and horizontal.
Reference
Cotran PR, Bajant AM. Congenital corneal opacities. Int Ophthalmol Clin 1992;32:93–105.
Developmental Disorders
Microcornea And Megalocornea
Summary
Small (horizontal diameter <10 mm, or 9 mm in a newborn) and large (horizontal diameter >13 mm) corneas, respectively.
Etiology
Undergrowth or overgrowth of the optic cup during development. Microcornea is generally autosomal dominant; megalocornea usually X-linked recessive.
Signs and Symptoms
Small-appearing (Figure 1.22A) or large-appearing (Figure 1.22B) eye.
Demographics
Megalocornea seen in male patients due to X-linked inheritance pattern.
Ophthalmic Findings
Refractive errors (hyperopia in microcornea, myopia in megalocornea), glaucoma, other ocular abnormalities.
Systemic Findings
 Microcornea is associated with myotonic dystrophy, fetal alcohol syndrome, Weill-Marchesani, Ehlers-Danlos, and Rieger syndromes, and Norrie disease.
Microcornea is associated with myotonic dystrophy, fetal alcohol syndrome, Weill-Marchesani, Ehlers-Danlos, and Rieger syndromes, and Norrie disease.
 Megalocornea is associated with Marfan syndrome, Down syndrome, mucolipidosis type II, craniosynostosis, facial abnormalities, dwarfism, mental retardation, and ichthyosis.
Megalocornea is associated with Marfan syndrome, Down syndrome, mucolipidosis type II, craniosynostosis, facial abnormalities, dwarfism, mental retardation, and ichthyosis.
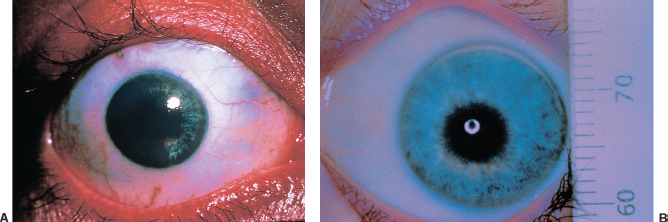
FIGURE 1.22. A: Microcornea. Microcornea is a descriptive term for a cornea with a diameter less than 11 mm. B: Megalocornea. Congenital enlargement of the cornea (and often the whole globe) with a diameter of almost 14 mm.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


