Chapter 76 White Spot Syndromes and Related Diseases
Introduction
The white spot syndromes (WSS) are a group of diseases characterized by inflammation and dysfunction of the outer retina, retinal pigment epithelium, choroid, or a combination of these. They often present a diagnostic and therapeutic challenge for clinicians and researchers. The etiologies of WSS remain unknown. This chapter discusses: birdshot chorioretinopathy (BCR), acute posterior multifocal placoid pigment epitheliopathy (APMPPE), serpiginous choroiditis, relentless placoid chorioretinitis, persistent placoid maculopathy, multifocal choroiditis and panuveitis (MFC), punctate inner choroidopathy (PIC), progressive subretinal fibrosis and uveitis syndrome (PSFU), multiple evanescent white dot syndrome (MEWDS), acute zonal occult outer retinopathy (AZOOR), acute idiopathic blind spot enlargement (AIBSE), and acute macular neuroretinopathy (AMN). Many entities are included in the differential diagnosis of these diseases. These include: granulomatous diseases, such as sarcoid, tuberculosis, sympathetic ophthalmia; masquerade syndromes like syphilis and intraocular lymphoma; infectious etiologies including toxoplasmosis, pneumocystis choroidopathy; and other entities such as presumed ocular histoplasmosis, and Behçet disease.1 In addition, in some cases, degenerative processes such as drusen may appear similar to the whitish-yellow lesions of WSS.
An autoimmune etiology has been hypothesized2 and proposed specifically for birdshot chorioretinopathy, acute zonal occult outer retinopathy (AZOOR), and multiple evanescent white dot syndrome (MEWDS).3 As yet, no characteristic pattern of antiretinal antibodies has been found. These entities need to be distinguished from entities with known neoplasias such as cancer-associated retinopathy (CAR) or melanoma-associated retinopathy (MAR). There are similar features including panretinal dysfunction, rapid progression, ERG changes, family history of autoimmune disease, retinal antibody activity on Western blot, and improvement in symptoms with immunosuppression. An increased prevalence of systemic autoimmunity in both patients with WSS and their first- as well as second-degree relatives, was found by Pearlman and colleagues.4 This suggests that this group of diseases occurs in families with inherited immune dysregulation that predisposes to autoimmunity.
It has also been suggested that several of these entities may be “related” or even represent a spectrum of the same process. Multifocal choroiditis (MFC), punctate inner choroiditis (PIC), MEWDS, acute macular neuroretinopathy (AMN), and AZOOR share commonalities and Gass referred to them as the AZOOR complex.5 These include female preponderance, zones of visual field loss that are usually contiguous to the blind spot, photopsias, and ERG changes such as reduced amplitudes. In addition, MFC and APMPPE have also been found in the same patient decades apart.6 Other diseases that have been noted in the same patient include MEWDS and AZOOR as well as MFC and PIC. This overlap may give support to a common underlying genetic predisposition.2
Birdshot chorioretinopathy
The term birdshot retinochoroidopathy was first used in 1980 by Ryan and Maumenee.7 It was a descriptive term for patients with multiple small, cream-colored fundus findings. These lesions are scattered around the optic disc and radiate to the equator in a “shotgun” pattern. Other terms such as vitiliginous chorioretinitis have been used.8 It has come to be known as birdshot chorioretinopathy (BCR) due to the histopathologic evidence that the primary lesions of the disease are in the choroid.9 It is a bilateral, chronic process with vitritis, retinal vasculitis, and cystoid macular edema (CME). Furthermore, it demonstrates the strongest link between any disease and any HLA class I antigen.10
Clinical course
Clinical symptoms
Patients present with complaints of blurred vision, floaters, and photopsias. Most have vision of 20/40 or better.9 The eye is generally not red or painful. Severe nyctalopia despite normal visual acuity may be a presenting symptom.11 Individuals also may describe an alteration in color vision12 or visual fields.13 Although Gass described a few patients who concurrently had vitiligo of the skin,8 BCR is thought to be a purely ocular disease.
Epidemiology
BCR is a rare chronic posterior uveitis. Shah and colleagues did an extensive review of English literature in 2005 and reported the following epidemiologic characteristics.9 Birdshot chorioretinopathy accounts for 0.6–1.5% of patients referred to tertiary centers for uveitis, or 6–7.9% of patients with posterior uveitis. There is a slight female predominance in the literature, at 54.1%. The mean reported age at the onset of disease is 53 years. It is generally not a disease of children. The oldest reported age at onset was 79 years. Patients are predominantly white; there have only been two reported exceptions. Finally, the HLA-A29 allele (which is present in about 7% of Caucasians) is strongly associated with BCR.14 The presence of this allele has been associated with a risk factor of 50–224 to develop BCR. Ninety percent or more of patients with BCR are HLA-A29-positive. Further sequencing of this allele has uncovered 11 subtypes. The HLA-A29*02 subtype is 20 times more prevalent in Caucasians than the HLA-A29*01 type, and the rest are exceedingly rare.15 HLA-A29*02 subtype is most commonly associated with BCR.
Fundus findings
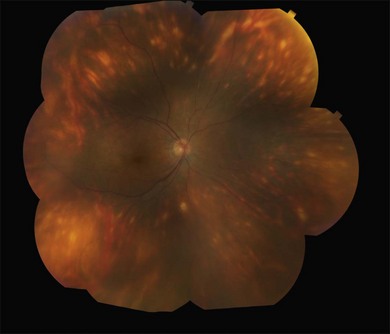
It is important to recognize that symptoms can precede the onset of the classic fundus findings by several years.16 There is a range of presentations of the lesions associated with BCR. These birdshot lesions can be oval or round in shape, typically about 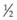 or
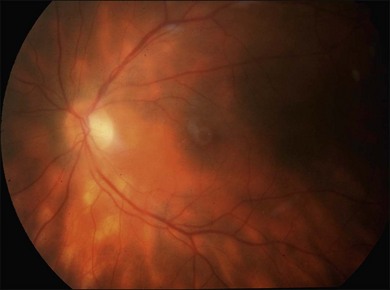
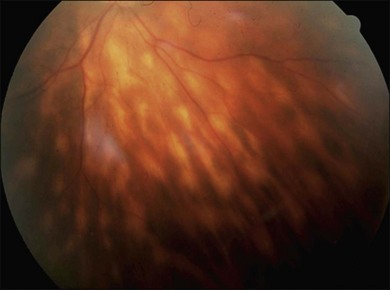
or 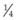 disc diameter in size. They appear deep to the retina (Fig. 76.1). They can be subtle, and asymmetric between eyes. The lesions can be distinct or poorly defined and can occasionally coalesce. They tend to cluster near the nerve and most commonly nasal and inferior to the disc.17,18 There is a pattern of linear radiation away from the nerve to the periphery7 (Fig. 76.2). They may appear to follow choroidal blood vessels peripherally. There is no hyperpigmentation or clumping noted. The retinal pigment epithelium (RPE) and overlying retina appear intact. There has been a report of these choroidal lesions preceding symptom onset in BCR, but this is not the norm.19
disc diameter in size. They appear deep to the retina (Fig. 76.1). They can be subtle, and asymmetric between eyes. The lesions can be distinct or poorly defined and can occasionally coalesce. They tend to cluster near the nerve and most commonly nasal and inferior to the disc.17,18 There is a pattern of linear radiation away from the nerve to the periphery7 (Fig. 76.2). They may appear to follow choroidal blood vessels peripherally. There is no hyperpigmentation or clumping noted. The retinal pigment epithelium (RPE) and overlying retina appear intact. There has been a report of these choroidal lesions preceding symptom onset in BCR, but this is not the norm.19
Other ocular findings
The anterior segment generally has minimal inflammation without prominent keratic precipitates. Fine keratic precipitates have been described in some series.18 Due to the mild nature of anterior segment inflammation, posterior synechiae do not occur. Signs of posterior inflammation include retinal vasculitis, optic disc edema, CME, and epiretinal membrane formation. Rhegmatogenous retinal detachment has been reported.7,20,21 It is unclear if this is related to this disease entity or to an association with uveitis. Other common findings include diffuse narrowing of the retinal arterioles, perivascular nerve fiber layer hemorrhages, and tortuosity of retinal vessels. Choroidal neovascularization (CNV) can occur.10,22,23 It has been postulated that this occurs due to the uveitic component causing CNV rather than ischemic factors.24,25
A consensus document on the diagnostic criteria for BCR was published in 2006.26 Required characteristics include: bilateral disease, presence of at least three peripapillary lesions inferior or nasal to the optic nerve in at least one eye, low-grade anterior segment inflammation (less than or equal to 1+ cells), and low grade vitreous inflammation (less than or equal to 2+ vitreous haze). Supportive findings include: HLA-A29 positive, retinal vasculitis, and CME. Finally, exclusion criteria include: presence of significant keratic precipitates, posterior synechiae, or diagnosis of infectious, neoplastic, or inflammatory disease that may cause multifocal choroidal lesions.
Clinical course and prognosis
The disease is chronic in nature and apparently does not regress. Again, symptoms of blurred vision, color deficiency, contrast sensitivity issues, and visual field problems may be present for years prior to the onset of fundus lesions. Visual acuity may be normal despite these complaints. It remains a poorly understood disease and no consensus on management and treatment has been found. Many patients have a slow decline in vision, despite treatment.9A final visual acuity of 20/40 or better in the best-seeing eye was reported in 75.1% of patients. However, 9.8% of patients were legally blind at follow-up (in the review of literature by Shah and colleagues9). Generally, macular edema was the commonest cause of visual decline in 50.5%.9 Choroidal neovascular membranes developed in 5% of eyes.22,23 These occur near the optic disc and can be bilateral. Finally, optic disc edema leading to atrophy can also affect visual prognosis.
Imaging
Fluorescein angiography
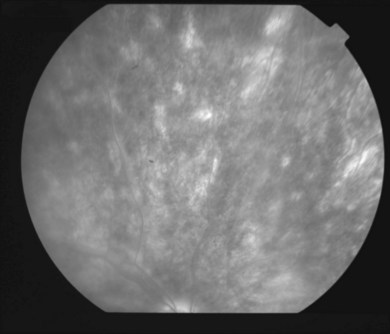
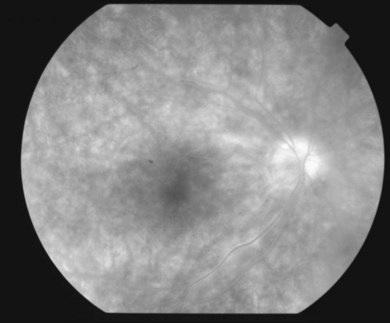
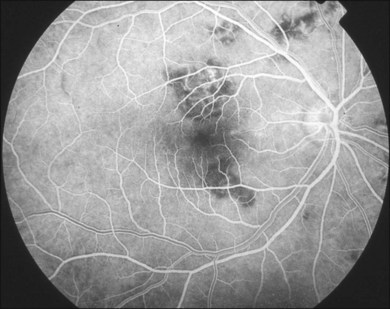
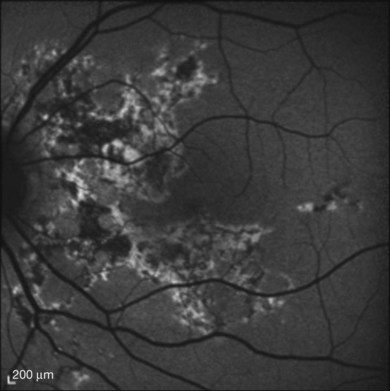
The creamy white spots have variable appearance on fluorescein angiography (FA) and the disease may be more evident clinically than with this mode of imaging (Fig. 76.3). The lesions can hypofluoresce in the early phase and there can be diffuse hyperfluorescence in the late phases (Fig. 76.4). One theory suggests that the lesions are likely in the outer choroid and associated with large choroidal vessels, thus many of the lesions show neither hypofluorescence or hyperfluorescence in any phase. The diffuse hyperfluorescence seen may represent a deep inflammatory focus that accumulates fluorescein.18 Angiographic findings may include increased transit time, leakage from retinal vasculature leading to CME, optic disc hyperfluorescence (Fig. 76.5), and disc or retinal neovascularization.8,27,28
Indocyanine green angiography
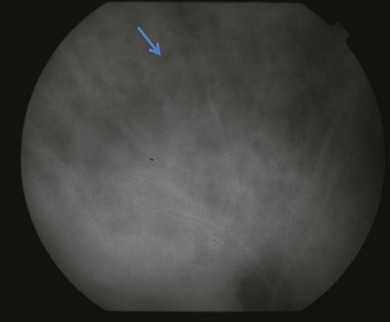
Indocyanine green angiography (ICGA) is an important diagnostic test. In the active disease,28 the birdshot lesions appear hypofluorescent during the intermediate phase of angiography and appear to be bordered by medium-to-large vessels28–30 (Fig. 76.6). The choroidal vessels appear indistinct. Late in the ICGA, there is diffuse choroidal hyperfluorescence. As with all the white spot syndromes, theories to explain this hypofluorescence include choroidal ischemia versus blockage from inflammatory infiltrates. However, in birdshot chorioretinopathy it is agreed upon that the site of the pathology is primarily the choroid. In the acute stages, the inflammatory infiltrates may be denser and block fluorescence. In late stages of the disease, it is thought that the lesions become more atrophic and choroidal vasculature may become more visible. The lesions can become more isofluorescent in this phase of the disease or they may remain hypofluorescent.27,28
Optical coherence tomography
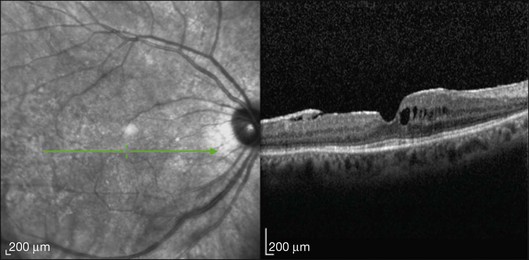
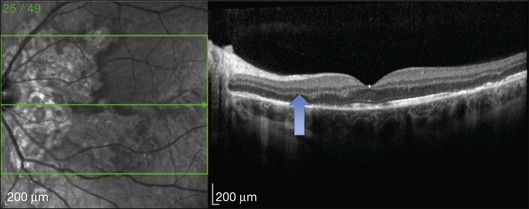
This mode of imaging has primarily been used to follow CME, which is the most common cause of vision loss in BCR31 (Fig. 76.7). Photoreceptor loss is not a prominent finding early on in the course.
Fundus autofluorescence
Giuliari and colleagues looked at fundus autofluorescence (FAF) of 18 eyes and found a spectrum of patterns.31 They found that hypoautofluorescent lesions on FAF did not necessarily correspond to clinical birdshot lesions. Commonly (15/18 eyes) more FAF lesions denoting RPE defects were seen than clinical lesions. Koizumi and colleagues looked at 8 patients. They found that some of these hypofluorescent lesions could be placoid in nature and involve the macula.32 In addition, a perivascular (retinal) linear hypofluorescent FAF pattern was noted by both groups of investigators.31,32 It was postulated that this finding may suggest that the retinal vessels may play a role in inflammatory-driven damage of the RPE and serve as a conduit for inflammatory factors. The observation that the clinical choroidal lesions did not always correspond to the FAF defects suggests that the choroid and RPE may be affected independently.32 Furthermore, RPE defects in the macula could also be another cause of vision loss in these individuals. FAF may be valuable in evaluating these patients as lesions may be clinically difficult to see.
Electrophysiology
Priem and coworkers also studied pattern-evoked cortical potentials and found abnormal findings in 53.3% of patients.33
Electroretinogram
Electroretinography (ERG) is an important test for following BCR. Abnormal electroretinograms (ERGs) were reported in 88.8% of patients.9 There appear to be two groups of patients: those who develop abnormal ERG early in the disease process and those that develop abnormal results late in the course.34 Early ERGs may demonstrate supernormal ERG amplitudes which may be related to retinal inflammation. In this stage, there may be a greater decrease in b-wave amplitude versus a-wave amplitude.21,34 This is a negative ERG pattern, and is not pathognomonic to BCR. It suggests that the Müller and bipolar cells are more affected than the photoreceptor–retinal pigment epithelium complex. This has been seen in autoimmune retinopathy. Rod dysfunction may occur before cone dysfunction: the rod b-wave may be affected prior to photopic b-wave and flicker response in most patients. In late phases of the disease, there is a progressive decrease in a- and b-wave amplitudes. ERG findings have been noted to improve with treatment.35 Some authors argue that the reversible nature of the ERG abnormalities suggest a non-ischemic etiology.15 Inflammation of the retinal vasculature could lead to inner retinal dysfunction, while choroidal inflammation could be the cause of altered outer retinal function.
Visual field testing
Visual field abnormalities are present in patients with BCR. These include: peripheral constriction, enlarged blind spot, central or paracentral scotomas, and generalized diminished sensitivity.8,17,18,20,33 It is not certain whether these visual field defects occur due to ganglion cell, optic nerve, or outer retinal dysfunction, but it is unlikely that the birdshot lesions themselves cause blind spots.9 Visual field abnormalities may be reversible with immunosuppression.36
Systemic associations
There are no definitive systemic associations. Shah and colleagues’9 review of the literature revealed hearing loss in some patients.8,37 As mentioned earlier, cutaneous vitiligo associated with BCR has also been reported in a few patients.8,18 In addition, Priem and Oosterhuis reported an increased incidence of vascular disease in their series. Of 102 patients, 16 had hypertension, 5 had coronary artery disease, 2 had a history of cerebrovascular accident, and 2 had a central retinal vein occlusion.18 These numbers are not impressive considering the age of the patients.
Pathogenesis
The pathogenesis of BCR is unknown. Inflammation appears to be a primary feature. The histopathological findings in a few eyes with BCR have been reported. These suggest that the spots may be related to accumulation of lymphocytes in the choroid at multiple levels, occasionally associated with hemorrhage.38 Some of the foci were adjacent to the choroidal vessels. The RPE, ciliary body, and iris were not involved. Some lymphocytes were found around the retinal blood vessels and in the optic disc. The lymphocytes were primarily CD8+T-lymphocytes. Inflammation is strongly associated with HLA-A29, which suggests a genetic predisposition to this disease. However the fact that HLA-A29 is present in 7% of the white population, and BCR is so rare tells us other factors are at work. Other genes are highly suspected. The more recent discovery of HLA class I-specific killer cell immunoglobulin-like receptors (KIR) led to epidemiological studies implicating KIR-HLA gene combinations in disease.39 The combination of KIR and HLA gene variants appears to increase the risk of developing BCR in HLA-A29-positive individuals. The presence of these in the absence of strong inhibition may activate natural killer cells and T cells against intraocular self-antigens thus inciting an autoimmune process. Familial history of autoimmunity is likely.2
Differential diagnosis
Birdshot chorioretinopathy can usually be distinguished from other disorders by history and physical findings. However, entities such as pars planitis, intraocular B-cell lymphoma, syphilitic chorioretinitis, sarcoidosis, sympathetic ophthalmia, and other white spot diseases, especially multifocal choroiditis and panuveitis syndrome, should be considered. Sarcoidosis (see Chapter 78, Sarcoidosis) and BCR may be the most difficult to distinguish from each other.9
Management/treatment
In the review of the literature by Shah and colleagues9 most cases were treated; however, no definitive guidelines for the initiation of treatment were given. This chronic progressive disease may not be sight-threatening in the early course, but macular edema with retinal damage, photoreceptor dysfunction, RPE atrophy, and optic nerve damage are the end results. Corticosteroids have been the mainstay of treatment. Oral, sub-Tenon’s, intraocular, and most recently sustained release fluocinolone acetonide40 have been used. Corticosteroids can reduce CME,7,21,41,42 inflammation,21 and optic disc edema.41 They have also been reported to decrease symptoms such as nyctalopia and issues with contrast sensitivity.11 Systemic steroids carry their own risks. Local steroid injections increase the risk of cataract and cause glaucoma with repeated dosing. Implantation of a fluocinolone acetonide sustained-release device has been shown to eliminate the need for systemic therapy, however it also has a high risk of cataract progression and glaucoma.40
Immunosuppressive therapy
Steroid-sparing agents have been used for long-term management of refractory cases. The three classes of immunosuppressives have been used. Cyclosporine has been used as it inhibits T lymphocytes and prevents S-Ag-induced experimental uveitis.10 Low-dose cyclosporine has proven to have positive visual effect in conjunction with steroids or alone.43 Nephrotoxicity is the primary side-effect and hypertension can be a problem.43 Antimetabolites such as azathioprine, mycophenolate mofetil, and methotrexate have been adjunctive or used in monotherapy. Side-effects of these drugs include bone-marrow suppression and hepatotoxicity.44 The use of the alkylating agents cyclophosphamide and chlorambucil has also been reported. Side-effects such as bone-marrow suppression and development of malignancies must be weighed for this class. Daclizumab, a monoclonal antibody against the alpha-subunit of the IL-2 receptor of T cells, has recently been found to have value in treating BCR.44,45 The long-term efficacy of these agents needs to be determined and weighed against potential side-effects.
Anti-VEGF therapy has been documented to be useful in treating CNV associated with inflammatory chorioretinal disorders.46
Placoid diseases
Acute posterior multifocal placoid pigment epitheliopathy
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) was first described in 1968 by Gass.47 He presented 3 healthy young female patients who developed the acute onset of bilateral central vision loss associated with multifocal placoid (plate-like) lesions at the level of the outer retina and the RPE, although he had also considered a primary choroiditis. Discussion regarding the site and etiology of the lesions would continue as the disease was further described in later years.48–57
Clinical course
Clinical symptoms
Patients present with rapid onset of central vision loss that may be described as blurred vision, paracentral scotoma, metamorphopsia, “spots” in the vision, and photopsias.58 Initial vision at presentation is 20/25 or worse in about 77% of eyes and 20/40 or worse in 58%.58 Deficits can be unilateral or bilateral (more common 75%).59 If unilateral, the second eye can become involved in a few days or weeks. Headaches, stiff neck, and malaise may accompany these ocular symptoms. A history of an antecedent viral syndrome or recent vaccination may be obtained.
Epidemiology
Males and females are equally affected and generally this occurs in young adults. Typically this presents between age 20 and 50 with the mean age of onset being 26.58,59 Recently, Taich and Johnson describe a syndrome resembling APMPPE in older adults (over age 50).60 These elderly individuals characteristically may have a worse outcome, with moderate or severe vision loss due to the development of geographic atrophy and CNV. These patients resemble the entity persistent placoid maculopathy (see below) and may not have APMPPE.
Fundus findings
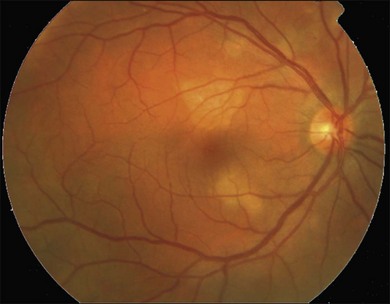
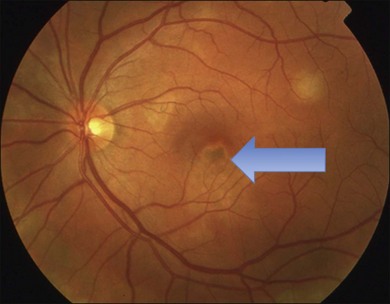
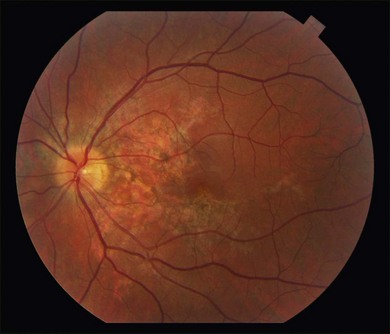
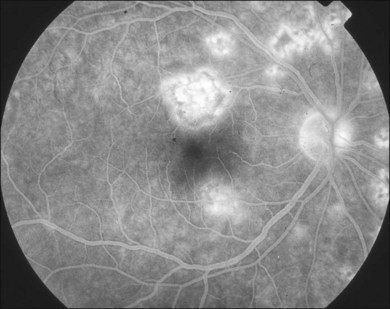
Gass described the presence of multiple round and confluent cream colored, flat lesions with indistinct margins scattered in the posterior pole. Lesions are not found anterior to the equator. The presence of these lesions is typically bilateral (Figs 76.8, 76.9). Fresh lesions can develop over the next few weeks, therefore lesions of differing ages can be visible. The placoid lesions tend to clear centrally initially leaving hypopigmentation. Later there is mild pigment mottling which develops into condensation of the pigment midperipherally, finally an increasing degree of coarse pigment clumping occurs (Fig. 76.10). These lesions can enlarge, but generally do not. In the original article, there was no description of serous macular detachment associated with APMPPE. However, later reports described localized serous retinal detachments over the lesions.52,55,61–65 This feature occasionally makes it difficult to distinguish APMPPE from Harada’s disease,62,66,67 and some have thought that these entities may form a spectrum of disease.68 It is more likely that there is an underlying common pathology that leads to the serous fluid.69 Gass commented on how remarkably the choroid and retina remain relatively intact during the course of the disease. However, Spaide described choroidal infiltrations in the periphery70 of a patient with acute APMPPE. In addition, multiple authors have described an association with retinal vasculitis71,72 and retinal vein occlusion has also been seen.71,72 Other findings can include subhyaloid hemorrhage72 and rare CNV.73 Case reports have also illustrated optic nerve involvement with disc edema.71,74
Other ocular findings
Although vitritis is not a significant component of APMPPE, the degree of ocular inflammation varies widely. An anterior uveitis75 and granulomatous anterior uveitis76 have been described. In addition, corneal stromal infiltrates77 have been mentioned.
Clinical course and prognosis
Generally there is improvement of the visual symptoms within 2–4 weeks. APMPPE has a relatively good prognosis when compared with other placoid white spot syndromes. However Fiore and colleagues58 reviewed the literature as well as a cohort from their own institution and showed that approximately 50% of patients have an incomplete recovery and 25% of patients have 20/40 vision or worse. Sixty percent of eyes have residual visual symptoms. Foveal involvement at presentation is an important prognosticator. Eighty-eight percent of eyes without foveal involvement proceed to full visual recovery in contrast to 53% of eyes that presented with foveal involvement. Unfortunately, approximately 70% of eyes present with foveal involvement. Photoreceptor involvement may ultimately limit visual prognosis. Progressive improvement of visual acuity usually follows the resolution of the lesions. Gass initially reported that visual recovery could continue on for months after the lesions resolved, even up to 6 months.47 Most visual recovery occurs within 1 month.49,51,52,55,58,61
Imaging
Fluorescein angiography
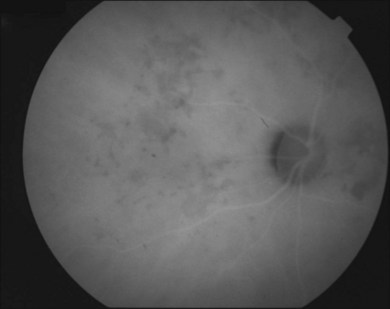
Gass47 described the lesions in the early phase as nonfluorescent (Fig. 76.11) and that the choroidal fluorescence is obstructed. Later in the angiogram there is a progressive, irregular staining of the lesions (Fig. 76.12). Ryan and Maumenee postulated that this could represent localization to the RPE or choriocapillaris rather than implying an infiltrate of the outer choroid.48 Gass also stated that the underlying choroidal circulation appeared intact below the healing lesions.47 However, later ICGA studies suggested that some choroidal hypoperfusion did exist.78,79 This is discussed below. As the process becomes inactive, hyperfluorescence corresponding to window defects in the RPE develops and staining is no longer evident (Fig. 76.13). There is a clear visibility of the large choroidal vessels in areas of confluent atrophy. However, this does not exist in all areas, implying some RPE remains intact.53 Either loss of the choriocapillaris or abnormal circulation in the confluent atrophied areas is suggested. In addition to these FA findings, there is also blocked fluorescence from pigment clumping.
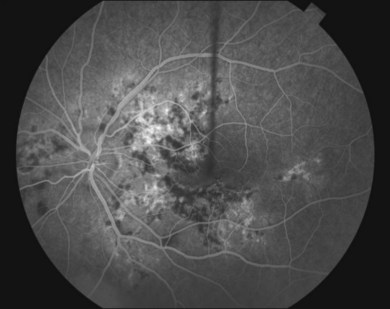
The retinal vessels and optic nerve appeared normal47,53 in original descriptions; however later reports described an association with retinal vasculitis, retinal vein occlusions, and disc edema.
Indocyanine green angiography
Since ICGA studies focus on the choroidal circulation, its findings in APMPPE have been important in developing theories of pathogenesis.79–84 Acute lesions show early hypofluorescence (Fig. 76.14). In the late phases, these lesions become more defined in shape. These areas are more numerous than the placoid lesions seen clinically. As the disease heals, the hypofluorescence in the late phase becomes less defined and smaller. This lends some support to the theory of choroidal ischemia as an underlying factor in the pathogenesis of APMPPE. It is postulated that there is more hypofluorescence in the acute phase due to the additional presence of swollen outer retina or RPE cells in response to choroidal ischemia (clinically presenting as placoid lesions). As these disappear, there is less blockage and thus less hypofluorescence. Photoreceptor damage may also play a role in this as well, as elucidated by OCT studies and discussed below. Studies show that the hypofluorescence in the late phases can also completely resolve,78 suggesting that choroidal vasculopathy, if present, may be a transient process.
Optical coherence tomography
Many studies have described the OCT findings in APMPPE.67,85–90 Garg and Jampol reported outer retinal abnormalities in APMPPE using time domain technology. A serous retinal detachment had reflective material within the subretinal fluid. It was postulated that this was either proteinaceous material or edematous RPE. There was rapid resolution of the serous detachment and the material disappeared.64
Lofoco and colleagues showed that in the acute phases the OCT revealed a mild hyperreflective area above the RPE in the photoreceptor layer corresponding to the placoid lesions. Later the OCT scan revealed a nodular hyperreflective lesion in the plane of the RPE with mild underlying backscattering. They theorized that the hyperreflective areas may indicate inflammatory tissue and inflammatory cells or the presence of ischemic edema in the outer retinal layers.90 As ultra-high resolution (UHR)-OCT developed, Scheufele and colleagues show disruption of the outer retina early in the disease (Fig. 76.15). RPE disruption occurs as the lesions heal.91 Backscatter of lesions was observed in the acute inflammatory phase in UHR as well. They found photoreceptor atrophy as the lesions began to heal and persistence of it post resolution. They suggested that the backscattering of acute lesions in the outer retina represents inflamed or damaged photoreceptor cell bodies. In addition to illustrating photoreceptor and RPE degeneration, spectral domain OCT has also shown that in some patients with fluid associated with the placoid lesions, that there may actually be accumulation of intraretinal fluid rather than an exudative retinal detachment.64,85
Fundus autofluorescence
FAF imaging is directed at the retinal pigment epithelial layer and is therefore especially useful in APMPPE. Several studies have described FAF findings67,87,89,92,93 (Fig 76.16).
Spaide compared angiographic findings with autofluorescence. He noted that the early hypofluorescence seen on angiogram did not match up with observable changes of the RPE and he suggested this means there are choriocapillaris perfusion defects. In the late phase of the angiogram, there was staining of some of the lesions. These late-staining lesions matched the size and shape of lesions seen in fundus autofluorescence. As the lesions resolved clinically, they became pigmented centrally with a depigmented halo. On autofluorescence, centrally there was intense hyperautofluorescence, and the depigmented halo was hypoautofluorescent, implying atrophy. He postulated there was a centripetal contraction of the placoid lesions that produced this appearance. He notes the autofluorescence changes lagged behind the clinical appearance. In addition, he found that choroidal abnormalities seemed more numerous on fluorescein and indocyanine green angiography. He concluded the RPE abnormalities were a result of the choroidal abnormalities.93
Electrophysiology
Although these functional studies are not essential in the diagnosis of APMPPE, they do emphasize the role of RPE involvement as described by Fishman and colleagues. The electroretinogram is normal to minimally subnormal. However, abnormal light:dark ratios have been documented on EOG, suggesting a diffuse RPE problem. Furthermore, the ERG and EOG abnormalities can normalize, suggesting that this can be a transient RPE problem.53
Systemic associations
APMPPE has been linked to CNS manifestations94–96 including cerebral vasculitis,97–103 meningo-encephalitis104 and stroke.105–109 Cases of APMPPE associated with CN VI palsy110 and transient hearing loss111 have also been cited. Headaches are a common symptom and APMPPE has mimicked migraine with aura.112 Although many patients give a history of viral illness, the symptoms of malaise and headaches may thus be more related to a widespread underlying vasculitis. Cerebrospinal fluid analysis has shown pleocytosis,113 which lends credence to this. The CNS associations are not benign and death, though uncommon, is possible.
Systemic vasculitis114 has been implicated in APMPPE and has been described in a P-ANCA-positive patient.115,116 Other associations with erythema nodosum,49,57 ulcerative colitis,117 thyroiditis,118 nephritis,119,120 and juvenile rheumatoid arthritis121 lead to an underlying immune mediated or inflammatory link. Other associations include granulomatous diseases such as: Wegener’s granulomatosis (granulomatosis with polyangiitis),122 pulmonary TB,123 sarcoidosis.97,124
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


