Pathology of the Retina
Gavin W. Roddy
Robert H. Rosa Jr.
CONGENITAL ABNORMALITIES
Retinal Coloboma
Colobomas arise from an incomplete fusion of the two sides of the optic cup afer the optic vesicle begins its in-vagination. Tis process begins with the invagination of the optic vesicle during week 4 of gestation and ends with the edges of the clef in the optic vesicle fusing during weeks 5 or 6.1 Normally, perfect fusion of the clef occurs at the point where mesodermal blood vessels destined to become the hyaloid vasculature enter to supply the developing ocular structures.
Colobomas are typically located in the inferonasal quadrant, but may be present anywhere. Retinal colobomas are actually defects in the neurosensory retina, retinal pigment epithelium (RPE), and choroid, because the retina and RPE arise from the two-layered optic vesicle as it invaginates, and the development of the choroid is contingent on the development of the prior two.
Retinal coloboma can be seen in renal coloboma syndrome, an autosomal dominant condition characterized by optic nerve dysplasia and renal hypodysplasia. Additional associated abnormalities include scleral staphyloma, small corneal diameters, microphthalmia, and optic nerve cyst.2
Histologically, a thin glial layer of undifferentiated retina is present, containing retinal vasculature. Small areas of dysplastic rosettes also may be present at the border of the coloboma as it transitions to normal-appearing retina with resumption of the underlying RPE and choroid.3
Retinal Dysplasia
Retinal dysplasia represents a disorganization of the lamellar architecture of the neurosensory retina and may be found in normal-appearing eyes, or associated with microphthalmia, trisomy 13 and 15, Walker-Warburg syndrome, or Norrie disease.4 A distinct clinical entity known as microcephaly, lymphedema, chorioretinal dysplasia (MLCRD) has also been described.5, 6 and 7 Elongated ciliary processes may be seen projecting to the area of dysplasia. Inherited retinal dysplasia has been described with either an autosomal dominant or X-linked inheritance pattern, and it often coexists with persistent fetal vasculature (persistent hyperplastic primary vitreous).8
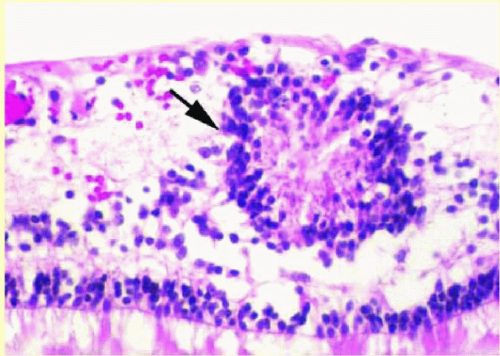 FIG. 13.1 Retinal dysplasia. A primitive rosette (arrow) composed of a group of cells arranged in a circle with a central neurofibrillary tangle. |
The presence of functional RPE is critical to normal retinal development, and any number of insults to the rapidly developing retina or RPE can lead to disorganization of the developing retina, subsequently causing rosette formation and retinal folds.9 Retinal dysplasia is a disorganization of nonneoplastic retina, unlike retinocytoma, which represents a benign neoplasm of the neurosensory retina. Rosettes (Fig. 13.1), similar to those seen in retinoblastoma, are commonly found in areas of retinal dysplasia, but have definite differentiating features from those occurring in retinoblastoma. Lahav and colleagues10
described four types of rosettes seen in retinal dysplasia with varying degrees of differentiation: (a) three-layer rosettes, (b) two-layer rosettes, (c) well-differentiated single-layer rosettes, and (d) primitive unilayer rosettes with undifferentiated retinal cells.
described four types of rosettes seen in retinal dysplasia with varying degrees of differentiation: (a) three-layer rosettes, (b) two-layer rosettes, (c) well-differentiated single-layer rosettes, and (d) primitive unilayer rosettes with undifferentiated retinal cells.
Myelinated Nerve Fibers
Myelinated (or medullated) retinal nerve fibers were described in 1856 as “chalk-white spots” by Virchow.11 Myelinated nerve fibers are seen in approximately 1% of all eyes and are usually present at birth and remain stationary, but rare cases may be acquired or progress.12, 13 and 14 Myelination generally emanates from the disk margin and clinically must be differentiated from the nerve fiber layer (NFL) edema seen with optic nerve head edema. It is generally unilateral, but may be bilateral in up to 7.7% of cases, and it can cause a visual field defect in the area of myelination that is typically not as depressed as would be expected for the degree of myelin obscuration of the deeper retina. Patients with myelinated retinal nerve fibers may be asymptomatic. It is often an isolated ocular finding or there may be coexistence of myopia, anisometropia, amblyopia, and strabismus.15 Myelin emanating from the disk is discontinuous with the myelin surrounding ganglion cell axons in the optic nerve, because ectopic oligodendrocytes are responsible for the NFL myelination.
Light microscopy shows normal-appearing retina except for eosinophilic thickening and occasionally increased cellularity within the retinal nerve fiber layer (RNFL). The glial cells appear different from the normalappearing astrocytes. They contain smaller, round, more basophilic nuclei—typical of oligodendrocytes.16
Perioral Abnormalities
The peripheral neurosensory retina is contiguous with the nonpigmented ciliary epithelium of the pars plana at the ora serrata. The ora serrata exhibits a more scalloped appearance nasally than temporally. Projections of neurosensory retina onto the pars plana (dentate processes) are most prominent nasally, occasionally with a retinal fold at their bases (meridional fold). Areas of pars plana projecting posteriorly between dentate processes (ora bays) may be enclosed and mimic a peripheral operculated retinal hole. Occasionally, dentate processes extend to the root of a ciliary process to form a meridional complex (Fig. 13.2). Cysts may also form in the pars plana, as well as oral pearls, which are small collections of PAS-positive material often with calcification and basophilia on hematoxylin-eosin (H&E) stain. Retinal tufts may also be visible at the perioral retina and consist of thin strands of glial and retinal tissue extending into the vitreous base; these may take on a cystic appearance (cystic retinal tuft).
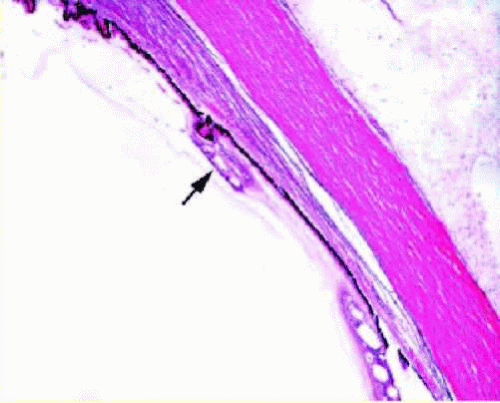 FIG. 13.2 Meridional fold. A focus of neurosensory retina (arrow) showing cystic change extends over the pars plana and is attached to a ciliary process. |
Lange Folds
Lange folds (Fig. 13.3) represent gross and microscopic artifactual peripheral neurosensory retinal detachment near the ora serrata that is present in infants and toddlers, generally less than 3 years of age. It is believed to be secondary to traction from the tightly adherent vitreous base and posterior lens zonular fibers on retina that has not fully developed adhesion to the underlying RPE. It does not represent an in vivo retinal detachment.17
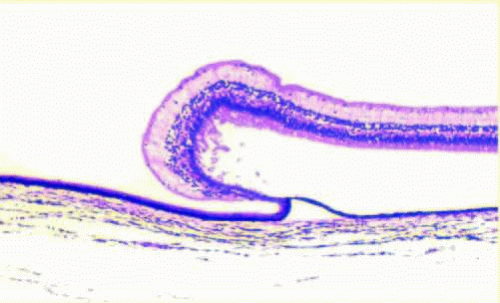 FIG. 13.3 Lange fold. An artifactual separation of the neurosensory retina with folding anteriorly onto the pars plana. |
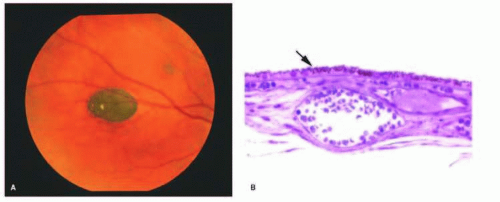 FIG. 13.4 A: CHRPE. Fundus photo of a pisciform (fish-shaped) hyperpigmented lesion of the RPE with sharp borders and depigmented lacunae. There is subtle attenuation of the RPE at the posterior (right) border of the lesion. B: Histology of CHRPE lesion showing abrupt transition (arrow) between hypertrophic RPE cells (right of arrow) and normal RPE (left of arrow) with apical distribution of melanin granules. |
Congenital Hypertrophy of the Retinal Pigment Epithelium
Congenital hypertrophy of the retinal pigment epithelium (CHRPE) is an abnormality in which the RPE cells are taller than normal, more numerous, and contain larger, round melanosomes, when compared with adjacent normal RPE.18, 19 Clinically (Fig. 13.4), the lesions are usually unilateral, have a sharp border, may contain depigmented lacunae that may enlarge or increase in number within the lesion with age, and may cause a relative or absolute scotoma on visual field testing.19 Generalized thickening of the RPE basement membrane often occurs beneath the areas of abnormal RPE. On microscopic examination, an absence of the RPE and photoreceptor layer with focal gliosis is observed in those areas seen clinically as hypopigmented lacunae. These lesions are now known to enlarge in diameter in up to 80% of cases20 and may rarely possess malignant potential.21
Grouped pigmentation (bear tracks) appears as multiple, small, grouped areas of hyperpigmentation at the level of the RPE. On light microscopy, an increase is noted in the number and size of melanin granules (usually of the normal ellipsoidal shape). The RPE cells in grouped pigmentation are of similar height compared with the normal surrounding RPE.22
Both CHRPE and grouped pigmentation are usually bilateral; however, in the setting of multiple, bilateral CHRPE lesions, an increased risk may be present of Gardner syndrome (Fig. 13.5) or familial adenomatous polyposis (FAP). Mutations in the adenomatous polyposis coli (APC) gene are responsible for FAP. The various extracolonic manifestations of FAP appear to localize to a specific location of the mutation on the APC gene. CHRPE mutations occur between codons 311 and 1444.23 CHRPE may be seen in up to 75% of patients with FAP.23 Multiple bilateral lesions have been reported to be a marker for FAP with a sensitivity of 84% and a specificity of 94%.24 Therefore, colonic screening in these patients may be warranted.25 Others have placed more emphasis on the general appearance of the lesion, with those lesions possessing irregular borders, a white tail of depigmentation on one margin, and presenting bilaterally, signifying an increased risk of colorectal cancer syndromes.26
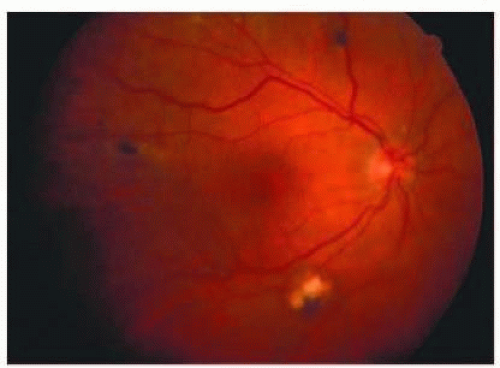 FIG. 13.5 CHRPE in Gardner syndrome. Fundus photo of a right eye with four visible CHRPE lesions. |
INFECTION AND INFLAMMATION
Posterior Segment Complications of HIV
A wide array of retinal pathology is directly related to infection with HIV, the development of AIDS, or the associated treatments. Approximately 50% to 75% of people worldwide with HIV/AIDS will manifest ocular complications of the disease, and at least 50% of these will be posterior segment complications.27 Highly active antiretroviral therapy (HAART) has drastically reduced HIV/AIDS-associated retinopathies in these patients.28, 29 The main categories of retinal pathology related to HIV/AIDS are microvascular, infectious, necrotic, and neoplastic.30 Recent reports have listed either cytomegalovirus (CMV)
HIV Retinopathy
HIV retinal microangiopathy/microvasculopathy is a microvascular disease that resembles diabetic retinopathy in terms of basement membrane thickening, pericyte loss, and endothelial cell abnormalities. These microvascular changes seen almost universally in histopathologic sections from patients with AIDS retinopathy are distinguished from HIV retinopathy, which is a clinical diagnosis referring to the presence of cotton-wool spots (CWS) from underlying ischemia.38 Between 40% and 60% of HIV-infected patients will at some time demonstrate CWS on examination.39, 40 and 41 CWS may be secondary to immune complex deposition within the vascular endothelium or endothelial cell infection with HIV.33 The prevalence of HIV retinopathy was once thought to be related to the degree of CD4+ depression,39, 42 but a more recent study attributed the development of HIV retinopathy to plasma HIV-1 RNA levels.33
Retinal Malignancy
Retinal malignancy in the setting of HIV/AIDS primarily consists of non-Hodgkin lymphoma (NHL). Intraocular NHL may manifest with choroidal infiltration, vitritis, or even necrotizing retinitis that is unresponsive to antiviral, antibacterial, and antiparasitic therapy.30 (See the “Primary Intraocular Lymphoma” section for further discussion.)
Immune Recovery Uveitis
Immune recovery uveitis (IRU) is an important cause of vision loss in an HIV-infected patient who is otherwise improving on HAART therapy. IRU is part of the spectrum of immune reconstitution inflammatory syndromes (IRIS) that is seen in the context of an opportunistic infection and following HAART-induced immune reconstitution. IRU is typically seen following CMV retinitis with increasing CD4+ T-cell counts (>100 cells per mm3).43 IRU is seen in approximately 20% of individuals who develop immune recovery.44 The disorder is characterized by transient or persistent vitritis, with subjective complaints of floaters or decreased vision, or by the presence of retinal neovascularization in the setting of inactive CMV retinitis in a patient who has had immune reconstitution with HAART. Other pathologic findings in IRU include cystoid macular edema, epiretinal membrane formation, and optic nerve neovascularization.45
CMV Retinitis
CMV retinitis occurs in patients with defective T-cell responses such as transplant recipients or patients with HIV/AIDS.46 Though the incidence of CMV retinitis has been reduced by approximately 75% to 90% with HAART therapy, it remains a significant cause of ocular morbidity in patients with HIV/AIDS34 and is a risk factor for increased mortality.47 Clinically, CMV retinitis occurs when profound depression takes place in the CD4+ cell count (<50) and takes one of three forms: fulminant retinitis with areas of hemorrhage (Fig. 13.6), granular retinitis, and most rarely, frosted-branch angiitis.48 Prior to HAART, the most common cause of profound vision loss (<20/200) was the spread of retinitis to involve the macula or optic nerve; however, with the increasing use of HAART, the primary cause of profound vision loss secondary to CMV is retinal detachment.49, 50 It is possible to discontinue suppressive anti-CMV therapy if HAART induces a persistent CD4+ count elevation (current recommended CD4 count >100 cells per µL).51, 52 and 53
 FIG. 13.6 CMV retinitis. Creamy areas of retinal necrosis and hemorrhage with vascular sheathing superiorly. |
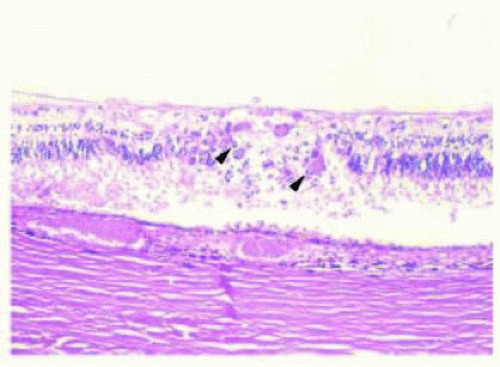 FIG. 13.7 CMV retinitis. H&E of an area of full-thickness retinal necrosis showing cytomegalo cells (arrowheads) with a nuclear “owl’s eye” appearance (intranuclear viral inclusions). |
Histopathologic examination of eyes with CMV retinitis shows necrotic neurosensory retina with choroidal infiltration by acute and chronic inflammatory cells and necrosis of the RPE (Fig. 13.7). Large cells with eosinophilic intranuclear and/or intracytoplasmic inclusions (cytomegalo cells) are typically present.39 The intranuclear inclusions may have an “owl’s eye” appearance.
Acute Retinal Necrosis and Progressive Outer Retinal Necrosis
Both acute retinal necrosis (ARN) and progressive outer retinal necrosis (PORN) occur as a result of either reactivation or primary infection with herpesviruses. PORN, also known as varicella zoster virus (VZV) retinitis or rapidly progressive herpetic retinal necrosis, occurs almost exclusively in patients with AIDS but may also rarely occur in other severely immunosuppressed individuals.54 In PORN, the vitreous and anterior segment are devoid of inflammation, and perivasculitis is generally not visible. In contrast, ARN, also known as Kirisawa uveitis, most classically occurs unexpectedly in immunocompetent individuals but may also occur in immunocompromised patients.30, 55, 56 and 57 ARN is most commonly caused by VZV or herpes simplex virus (HSV) or rarely by CMV or Epstein-Barr virus (EBV), and is associated with peripheral necrotizing retinitis, vitritis, iritis, and retinal arteriolitis. Histologically, full-thickness retinal necrosis occurs with coexisting inflammation of the RPE and choroid in both ARN and PORN, despite the apparent inner retinal sparing seen clinically with PORN.30 The incidence of retinal detachment in ARN is reported to be upwards of 70%, with blindness secondary to bilateral retinal detachment in 59% of affected patients.58, 59
Bacterial and Fungal Retinopathy
Septic retinopathy generally occurs via the hematologic dissemination of septic microemboli, often from bacterial endocarditis, although any episode of septicemia may lead to septic retinopathy. The classic clinical sign of bacterial embolization or infection is that of a whitecentered hemorrhage, called a Roth spot. Histologically, microemboli contain inflammatory cells, especially neutrophils, and bacterial or fungal organisms (Fig. 13.8). Leukemic retinopathy may also present with a similar clinical picture.
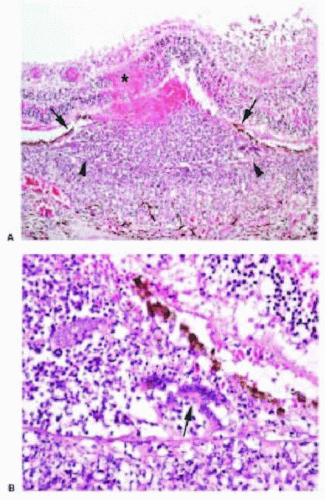 FIG. 13.8 Retinal abscess. A: H&E showing Bruch membrane (arrowheads) with overlying disrupted and elevated RPE (arrows) and intraretinal hemorrhage with necrosis (asterisk) and surrounding acute inflammatory infiltrate. B: Higher power H&E showing multinucleated giant cells (arrow) between Bruch membrane and the RPE. |
Although a host of fungi have been reported to cause fungal retinopathy, the two most common are Candida and Aspergillus.60 Candida septicemia is the most common cause of Candida retinopathy; however, the
likelihood of having Candida retinopathy after a single positive blood culture in the absence of any visual symptoms is exceedingly low.61 On the other hand, Candida retinopathy in the setting of true candidemia (>1 positive blood culture) carries a significant risk of mortality given the severity of the underlying infection. Aspergillus retinopathy usually occurs following solid organ transplantation with accompanying immunosuppression.62 The presentation is usually one of deep retinal and choroidal infiltration, occasionally presenting with a pseudohypopyon.
likelihood of having Candida retinopathy after a single positive blood culture in the absence of any visual symptoms is exceedingly low.61 On the other hand, Candida retinopathy in the setting of true candidemia (>1 positive blood culture) carries a significant risk of mortality given the severity of the underlying infection. Aspergillus retinopathy usually occurs following solid organ transplantation with accompanying immunosuppression.62 The presentation is usually one of deep retinal and choroidal infiltration, occasionally presenting with a pseudohypopyon.
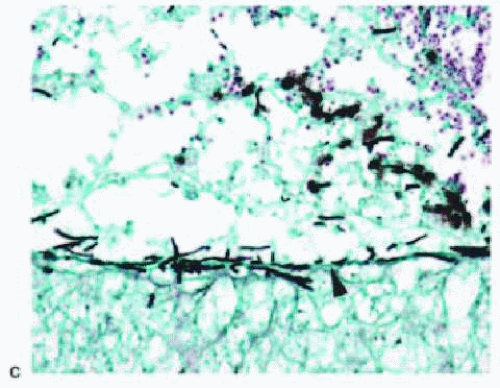 FIG. 13.8 (Continued) C: Gomori-methenamine-silver (GMS) stain showing fungal hyphae in the region of Fig. 13.8B. Note the localization of the fungal hyphae to the region of Bruch membrane. (arrowhead). |
DEGENERATIONS
Age-Related Macular Degeneration
Age-related macular degeneration (AMD) is likely the result of genetic predisposition and the additive effects of chronic injury to the RPE, Bruch membrane, and choriocapillaris as the result of oxidative stress, inflammation, and metabolic end-product deposition.63 These processes lead to the clinical spectrum observed in AMD, namely drusen, RPE hyperplasia or hypertrophy, RPE hyperpigmentation or hypopigmentation, geographic atrophy (GA), choroidal neovascularization (CNV), and disciform scarring.64 To date, the 1q3265, 66 and 10q2667, 68 loci are the most strongly associated with AMD. The 1q32 locus encodes complement factor H (CFH), while the 10q26 locus encodes the genes and Pleckstrin homology domain containing family A (PLEKHA1), age-related maculopathy susceptibility 2 (ARMS2), and HTRA serine peptidase 1 (HTRRA1).69 AMD is divided into two types: the nonexudative (dry) form, which afflicts about 85% to 90% of all patients with AMD, and the exudative (wet) form, which accounts for the remaining 10% to 15% of patients. Exudative AMD accounts for approximately 75% of the legal blindness due to AMD, and nonexudative AMD (GA) accounts for the remaining 25%.70, 71 It has been suggested that RPE dysfunction and loss is the inciting factor in both exudative and nonexudative
AMD.72,73
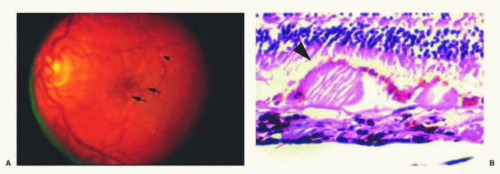 FIG. 13.9 Drusen. A: Fundus photograph displaying multiple soft drusen (arrows) and rare hard drusen (arrowhead). B: H&E of drusen demonstrating an eosinophilic, hyaline-like deposit with focal RPE attenuation (arrowhead) at the apex of the druse. |
The hallmarks of nonexudative AMD are drusen, GA, and RPE hypertrophy/hyperplasia. Drusen represent the earliest clinical sign of AMD and appear microscopically as focal, sub-RPE deposits of eosinophilic, hyalinelike material in the inner aspect of Bruch membrane (Fig. 13.9). Clinical descriptions, such as “hard” or “sof” drusen, do not easily translate to the histologic appearance
of these deposits. These deposits appear eosinophilic with H&E staining and are periodic acid-Schiff (PAS)-positive; moreover, the deposits may appear granular on light microscopic examination with focal areas of hyaline-like material within. Deposits in Bruch membrane and drusen contain complement proteins and various oxidation products,74 which may act as a stimulus for chronic unresolved inflammation.75 Factor H, a major inhibitor of the alternative complement pathway, was found to be synthesized by the RPE and accumulate within drusen in the RPE/choroid complex.65 Overlying RPE attenuation is often present and, occasionally, RPE cell migration within the subretinal space and/or pigment deposition occurs within the photoreceptor layer.76
of these deposits. These deposits appear eosinophilic with H&E staining and are periodic acid-Schiff (PAS)-positive; moreover, the deposits may appear granular on light microscopic examination with focal areas of hyaline-like material within. Deposits in Bruch membrane and drusen contain complement proteins and various oxidation products,74 which may act as a stimulus for chronic unresolved inflammation.75 Factor H, a major inhibitor of the alternative complement pathway, was found to be synthesized by the RPE and accumulate within drusen in the RPE/choroid complex.65 Overlying RPE attenuation is often present and, occasionally, RPE cell migration within the subretinal space and/or pigment deposition occurs within the photoreceptor layer.76
Deposited material has been further subdivided by electron microscopy into basal laminar and basal linear deposits. Basal laminar deposit is characterized by its location between the RPE plasma membrane and RPE basement membrane. It has a characteristic granular, PAS-positive appearance on light microscopy and exhibits widely spaced collagen bundles on electron microscopy. Basal laminar deposit represents RPE dysfunction and is correlated with clinical pigmentary changes. Basal linear deposit is characterized by its location within the inner collagenous zone of Bruch membrane, and its vacuolated, vesicular appearance on electron microscopy77 is due to its lipoprotein composition.78
GA is represented by well-circumscribed depig-mentation in the macular region secondary to RPE loss (Fig. 13.10). It is the most common cause of legal blindness in nonexudative AMD. Histologically, GA is identifed by attenuation or loss of the RPE and concomitant loss of the overlying photoreceptor cell layer and underlying chorio-capillaris (Fig. 13.11). At the edge of the area of GA, lipo-fuscin-flled RPE cells are often visible.64 Tere appears to be a linear relationship between the loss of RPE and the choriocapillaris in GA. Loss of the RPE is thought to occur first; it is thought that resultant hypoxia from vascular dropout may lead to future vascular endothelial growth factor (VEGF) production and CNV.73 At the other end of the spectrum, RPE hypertrophy or hyperplasia in the setting of AMD is seen on clinical and histologic examination as focal areas of subretinal or outer retinal pigmented cell accumulation.76
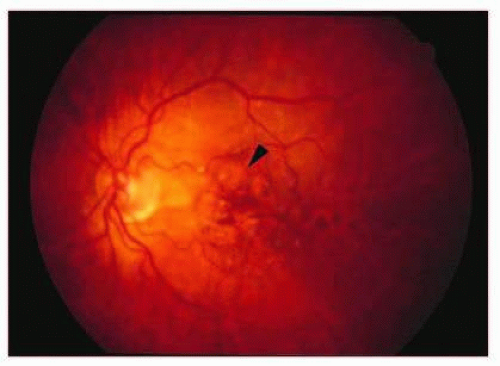 FIG. 13.10 GA. Fundus photo showing areas of focal RPE loss/atrophy (arrowhead). Note that this is a later fundus photo of the same eye seen in Figure 13.9A demonstrating the evolution of geographic RPE atrophy in areas containing large soft drusen. |
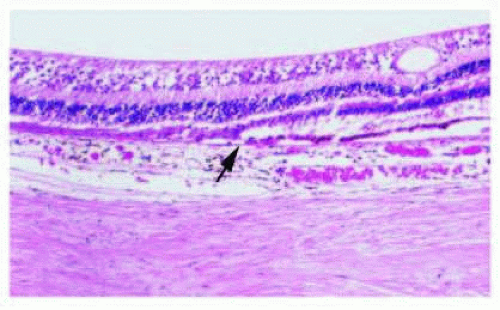 FIG. 13.11 GA. H&E showing abrupt cessation of the RPE (left of arrow) with loss of the overlying photoreceptor layer and attenuation of the underlying choriocapillaris. |
Exudative AMD may occur primarily, or secondarily from preexisting nonexudative AMD. In the setting of nonexudative AMD, drusen size, extent, presence of numerous soft drusen, and RPE hyperplasia have all been shown to increase the likelihood of progression to exudative AMD.79 Exudative AMD is characterized by the growth of new choroidal blood vessels, or CNV, into and through Bruch membrane and beneath either the RPE (Gass type I) or neurosensory retina (Gass type II) (Fig. 13.12).80 Degenerative changes in Bruch membrane, combined with inflammation and other stimuli for new vessel growth (particularly VEGF) allow for the growth of CNV through Bruch membrane into the subretinal space.64, 81 These layers of new vessels are often accompanied by inflammatory cells, multinucleated giant cells (Fig. 13.13), and macrophages.82, 83 Nets of new vessels leak fluid and are predisposed to bleeding, both of which contribute significantly to vision loss. Although the exact mechanism of this process is unknown, VEGF is known to play a significant role.84 As mentioned above, as GA leads to atrophy of the choriocapillaris and resultant hypoxia,
VEGF production may be increased in a compensatory manner. Additionally, as the RPE becomes dysfunctional causing drusen accumulation, VEGF may be produced owing to the infammatory stimulus.84 Te RPE produces VEGF in response to complement and oxidative stress.72 Te use of anti-VEGF therapies in exudative AMD has been a success story in the development of mechanism-based therapies.85
VEGF production may be increased in a compensatory manner. Additionally, as the RPE becomes dysfunctional causing drusen accumulation, VEGF may be produced owing to the infammatory stimulus.84 Te RPE produces VEGF in response to complement and oxidative stress.72 Te use of anti-VEGF therapies in exudative AMD has been a success story in the development of mechanism-based therapies.85
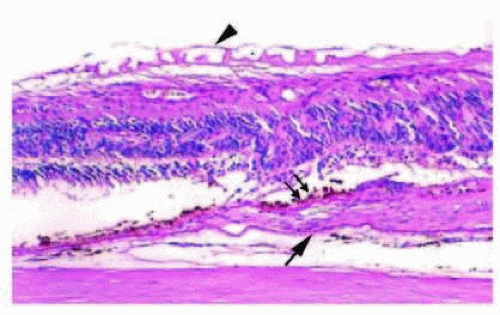 FIG. 13.12 Choroidal neovascularization (CNV). Fibrovascular tissue is present between Bruch membrane (arrow) and the RPE (double arrows). Note the epiretinal membrane (ERM) with ILM contraction features (arrowhead). |
Large, soft, confuent drusen can give rise to the clinical picture of an RPE detachment from the aggregation of drusenoid material and proteinaceous debris under the RPE. This form of RPE detachment is not secondary to CNV, so it is termed “drusenoid” RPE detachment. This lesion is more likely to progress to GA than to CNV.86, 87
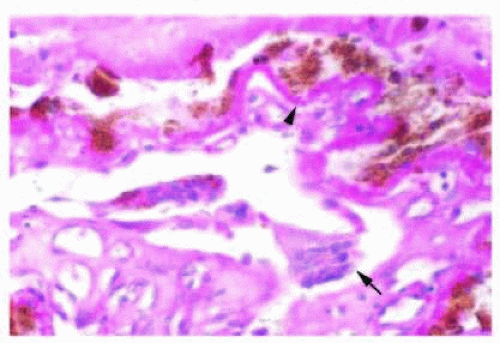 FIG. 13.13 Choroidal neovascularization. Irregular RPE is seen overlying basal laminar deposit (arrowhead) with underlying CNV. Multinucleated giant cells (arrow) are present, suggesting an inflammatory component to the neovascular membranes in AMD. |
Disciform scarring represents the end stage of exudative AMD. It commonly follows hemorrhage from CNV. The scar may have varying degrees of vascularization, and it may be located in the subretinal space, the sub-RPE space, or within Bruch membrane.77
Retinal angiomatous proliferation (RAP) is a variant of exudative AMD in which the new blood vessel growth arises from the retinal circulation, rather than from the choroidal circulation. It has long been known that retinal-to-choroidal anastomoses (RCAs) occur in late (disciform) stages of AMD; however, more recently, retinal neovascularization preceding CNV in the setting of exudative AMD has been described.77, 88, 89 Yannuzzi and coworkers90 first classified these RAPs into one of three subtypes on the basis of their suspected progression: (1) intraretinal neovascularization, (2) subretinal neovascularization, and (3) CNV. Histologically, these lesions appear as a fibrovascular network, with feeding arterioles and draining venules from the neurosensory retina, and may later appear as a frank RCA.89 (Please refer to Volume 3, Chapter 23 for a detailed discussion of AMD.)
Myopic Degeneration
Myopic degeneration generally does not occur with myopia of less than 6 to 8 diopters, and becomes increasingly common with higher degrees of myopia. Myopic degeneration is characterized by peripapillary retinal atrophy, focal linear breaks in Bruch membrane (lacquer cracks), RPE abnormalities (including focal pigmentation or Fuchs spot), peripheral retinal degeneration (especially cobblestone degeneration and lattice degeneration), and CNV.91 In extreme circumstances, posterior bowing of the globe occurs secondary to scleral thinning and results in a posterior staphyloma.92 With the number of high myopes increasing over the last few decades, pathologic myopia is becoming a greater cause of ocular morbidity.93
Angioid Streaks
Angioid streaks represent discontinuities in Bruch membrane that usually emanate from the optic disk or may exhibit a peripapillary ring configuration. Angioid streaks have been described in association with Paget disease of bone, Eales disease, pseudoxanthoma elasticum, sickle cell disease, and hemolytic anemias, but are most often idiopathic. Histologically, angioid streaks are focal breaks in a thickened and calcified (more basophilic) Bruch membrane. The overlying RPE and underlying choriocapillaris appear normal initially, but late CNV94 often
occurs. Parafoveal and subfoveal CNV in the setting of angioid streaks has been treated with photodynamic therapy (PDT).95, 96
occurs. Parafoveal and subfoveal CNV in the setting of angioid streaks has been treated with photodynamic therapy (PDT).95, 96
Central Serous Chorioretinopathy
Central serous chorioretinopathy (CSCR) is an idiopathic condition in which serous fluid accumulates under the neurosensory retina and occasionally the RPE.97 Previously, the primary pathology was thought to occur in the RPE, in which either a focal leak or break occurs in the outer blood-retinal barrier or a reversal of polarity in the RPE pump that normally provides for the net vitreal-to-choroidal flow of fluid. More recently, investigators have suggested that choroidal hyperpermeability is the cause of the retinal and RPE detachments in this disorder. The subretinal fluid may contain fibrin, which can appear whitish on examination and may predispose to a poor visual outcome.98
A recent case-control study confirmed and/or identified systemic steroid use, pregnancy, antibiotic use, alcohol use, untreated hypertension, and allergic respiratory disease as risk factors for CSCR.99 Though CSCR resolves without treatment in approximately 90% of cases, several different treatment modalities may be considered as CSCR may recur in up to 41% of patients100 and treatment may decrease the risk of recurrence.100, 101 and 102 Several treatment modalities have been employed,103, 104 including focal laser photocoagulation,105 PDT,105 and anti-VEGF injections.106 Long-standing CSCR may progress to cystoid degeneration of the retina and poor final visual acuity.107
Polypoidal Choroidal Vasculopathy
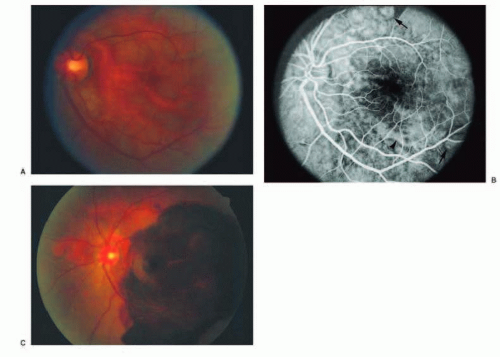 FIG. 13.14 PCV. A: Fundus photo showing red-orange, polypoidal and tubular lesions in the peripapillary and central macular region. B: Midphase FA demonstrating polypoidal (arrows) and tubular (arrowhead) hyperfluorescent lesions without apparent leakage. C: Fundus photo of the same patient showing marked submacular hemorrhage with persistence of the red-orange lesions in the peripapillary and region. |
Polypoidal choroidal vasculopathy (PCV),108 previously described as posterior uveal bleeding syndrome109 and multiple recurrent serosanguineous RPE detachment,110 is a disorder in which dilated, thin-walled vascular channels, apparently arising from the short posterior ciliary arteries, penetrate into Bruch membrane.111, 112 Ophthalmoscopically, the polypoidal lesions appear as elevated, red-orange, polyp-like or tubular subretinal lesions that are often associated with serosanguineous detachments of the retina and RPE (Figs. 13.14 and 13.15). These lesions may simulate hemorrhage from CNV associated
with AMD. In Japanese and Chinese patients, it primarily affects men (73% to 83% of the time) and is generally unilateral (83% to 87%),113, 114 whereas in Europe and the Americas it preferentially affects darker-pigmented patients (79%) with a female preponderance (96%), is more often bilateral (68%), and may be associated with hypertension (43%).111 More recently, polypoidal lesions have been identified in Caucasian patients, most of whom also have exudative AMD.115, 116
with AMD. In Japanese and Chinese patients, it primarily affects men (73% to 83% of the time) and is generally unilateral (83% to 87%),113, 114 whereas in Europe and the Americas it preferentially affects darker-pigmented patients (79%) with a female preponderance (96%), is more often bilateral (68%), and may be associated with hypertension (43%).111 More recently, polypoidal lesions have been identified in Caucasian patients, most of whom also have exudative AMD.115, 116
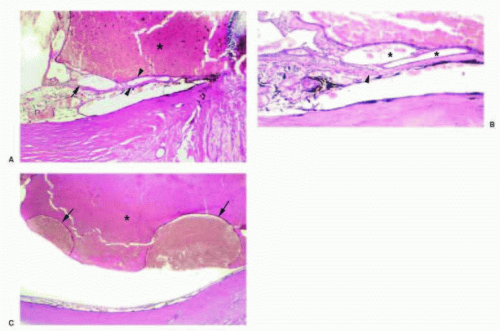 FIG. 13.15 PCV. A: Hemorrhagic retinal detachment (asterisk) and cavernous vascular channels (arrow) interposed between the RPE and the outer aspect of Bruch membrane (between arrowheads). B: Thin-walled vascular channels (asterisk) within the sub-RPE space overlying the outer aspect of Bruch membrane (arrowhead). C: Hemorrhagic RPE detachments (arrows) with overlying serosanguineous retinal detachment (asterisk). |
Peripheral Cystoid Degeneration
Peripheral cystoid degeneration can be subdivided into two separate entities, each with its own clinical and histologic appearance: typical peripheral cystoid degeneration (TPCD) and reticular peripheral cystoid degeneration (RPCD).
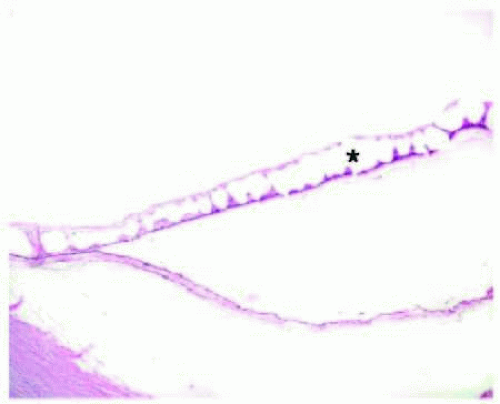 FIG. 13.16 TPCD. Peripheral cystic spaces (asterisk) in the neurosensory retina with coalescence to form schisis cavities. Müller cells span the space between the inner and outer retinal layers. The neurosensory retinal detachment is artifactual. |
TPCD is characterized by visible microcysts that, on fusing, form an irregular, lobulated, vessel-like channel. TPCD is generally located near the ora serrata, with a smooth or rounded posterior border. RPCD is most often found at the posterior border of TPCD and is usually bordered posteriorly by the retinal vessels. RPCD has a finer vascular pattern visible within the degenerative area, whereas TPCD usually obscures all but the most prominent retinal vessels.117, 118 Müller cells and other neural fibers provide the spanning support between the inner and outer retinal layers in both TPCD (Fig. 13.16) and RPCD,119 and both patterns of degeneration may progress
to allow the cysts to fuse and form larger areas of splitting within the retina (retinoschisis).
to allow the cysts to fuse and form larger areas of splitting within the retina (retinoschisis).
Retinoschisis
Retinoschisis is defined as a cleavage of the retina into an inner and an outer layer. It is most commonly seen as either typical or reticular degenerative retinoschisis, the result of the coalescence of lacunae in either TPCD or RPCD. Typical retinoschisis shows a splitting of the retina within the outer plexiform layer, whereas reticular retinoschisis demonstrates a splitting within the NFL. Retinoschisis arising from TPCD is generally flat, whereas retinoschisis arising from areas of RPCD has a higher likelihood of taking on a bullous appearance, progressing more posteriorly, and having both inner and outer layer holes.118 Treatment is generally not indicated unless there is a progression of the splitting to involve the macula, an associated nonrhegmatogenous retinal detachment (fluid from the schisis cavity passes under the retina through an outer leaflet hole), or an associated rhegmatogenous retinal detachment (liquefied vitreous gains access to the subretinal space from opercula within the inner and outer layers).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


