Age group (40–60 years)
Serpiginous choroiditis
Birdshot retinochoroidopathy
Age group (30–40 years)
APMPPE
MEWDS
PIC
MCP
Unilateral
MEWDS
Bilateral
APMPPE
Birdshot retinochoroidopathy
PIC
MCP
Small lesions (25–100 um)
PIC, MEWDS, ARPE
Large lesions (200 um and larger)
APMPPE
Serpiginous choroiditis
Subretinal fibrosis
Histopathology
The dots appear as a small granuloma which is composed of lymphocytes and macrophages. The lesions may occur in the choroid between Bruch’s membrane and the retinal pigment epithelium (RPE) or between the RPE and photoreceptors.
Serpiginous Choroiditis
Inflammation is seen as a gray-white fluffy choroiditis that occurs at the level of the RPE, choriocapillaries, and choroid [1]. Other names used to refer to serpiginous choroiditis include choroidal sclerosis, geographic choroiditis [2], geographic helicoid peripapillary choroidopathy [3], and helicoid peripapillary chorioretinal degeneration. It affects healthy individuals in their fourth to sixth decades of life although disease in younger individuals has been reported in the Indian population [4].
Etiology
Symptoms
Presentation may be with blurred vision, metamorphopsia, or central and paracentral scotoma. Visual loss depends on the involvement of the macula.
Clinical features depend on the type of serpiginous choroiditis.
Classification
Peripapillary [8] is the typical form and the active stage has a characteristic geographic pattern radiating outward in a serpentine fashion (Fig. 18.1). This centrifugal spread may involve the macula. Recurrences are common and occur from the edges of the previous scars. Lesions are self-limiting. Chronic inflammation can cause pigmentary clumping and atrophic patches in the retina with visibility of choroidal vasculature.
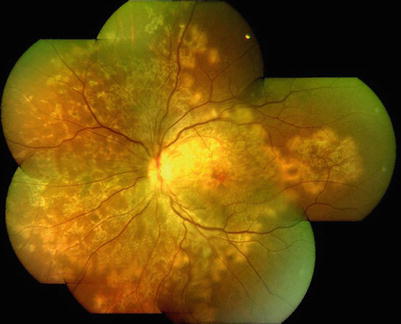
Fig. 18.1
Active serpiginous choroiditis
Macular serpiginous [9]
The macular variant of serpiginous choroiditis causes early symptoms due to foveal involvement or due to complications such as CNVM (Fig. 18.2).
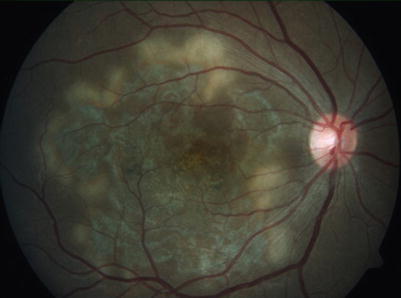
Fig. 18.2
Macular serpiginous choroiditis
Ampiginous choroiditis
Lesions in ampiginous choroiditis [10] appear to represent an intermediate between serpiginous choroiditis and APMPPE. They are bilateral and share similar angiographic features to serpiginous choroiditis and are multifocal as in APMPPE. Ocular tuberculosis forms the essential differential diagnosis in endemic areas.
Fundus fluorescein angiography [11] has the appearance of blocked fluorescence during the early phase as a result of combination of RPE cell edema and nonperfusion of choriocapillaries. In the later phase, the hyperfluorescence is because of tissue damage.
Indocyanine green angiography [12] detects subclinical lesions and is more useful as it delineates choroidal circulation better. The lesions appear hypofluorescent throughout.
Fundus autofluorescence [13] shows a discrete hypofluorescence accompanied by a hyperfluorescent halo in the active stages. After resolution a hypofluorescent edge is seen followed by a granular pattern of the lesion and finally a well-defined hypofluorescence.
OCT shows a disruption of the outer retinal architecture, with loss of the junction of the inner and outer segments of the photoreceptors and variable thinning/thickening of the RPE-Bruch’s membrane layer, corresponding to the scars.
Visual field plotting shows absolute or relative scotomas in areas of retinal lesions. An Amsler grid can be used for self-monitoring to help detect recurrence.
Electrophysiologic studies are usually normal.
Differential diagnosis
The most frequently considered differential diagnosis is tubercular serpiginous-like choroiditis [16]. However, these lesions have more vitritis and the multifocal lesions lie adjacent to retinal vessels. Investigations are required to confirm the diagnosis. APMPPE, multifocal choroiditis with panuveitis, and outer retinal toxoplasmosis may also need to be considered while making a diagnosis.
Complications
Direct involvement of the macula by an active lesion can cause visual loss. In the healed stages, choroidal neovascular membranes or scarring can occur. The most common cause of visual loss is CNVM that develops as a result of recurrent attacks. Less frequent complications are branch retinal vein occlusion, periphlebitis, serous retinal detachment, cystoid macular edema, or subretinal fibrosis.
Investigations
Complete blood count, Mantoux test, syphilis serology serum angiotensin-converting enzyme, and chest radiographs need to be done in all patients. Polymerase chain reaction (PCR), QuantiFERON-TB Gold test [17], nested PCR [18], or real-time PCR [19] can be done to confirm the diagnosis and to exclude infective and systemic causes of posterior uveitis. They can be performed on aqueous [20] or vitreous samples and chorioretinal biopsies.
Management
Factors to be considered are the activity of the lesion and macular involvement. Long-term follow-up is required to monitor response to treatment and to detect recurrence.
Systemic corticosteroids
Oral prednisolone in the dose of 1 mg/kg/body weight can be given alone or in combination. In patients with macular involvement, intravenous methyl prednisolone is administered in the dose of 1 g for 3 consecutive [21] days to hasten the resolution of serpiginous choroiditis.
Intravitreal triamcinolone acetate [22] is used in cases where systemic corticosteroids are contraindicated or in secondary CNVM.
Immunosuppressives: Azathioprine [23] is used at 1.5–2 mg/kg per day in isolation or in combination with oral prednisolone in the dose of 1 mg/kg. Methotrexate, mycophenolate mofetil, or cyclosporine [24] can be used as an alternative to azathioprine.
Triple-agent therapy [25]
The current management modality is a combination of cyclosporine, azathioprine, and prednisolone.
Alkylating agents and antimetabolites
Cyclophosphamide and biological therapy such as interferon alpha-2a [26] are other treatment options.
CNVM can be treated with argon laser, photodynamic therapy, or anti-VEGF therapy [27].
Acute Posterior Multifocal Placoid Pigment Epitheliopathy (APMPPE)
APMPPE is an inflammatory chorioretinopathy which was first described by Gass in 1968 [28]. Acute posterior multifocal placoid pigment epitheliopathy is a bilateral disease in otherwise healthy young adults and affects women and men equally. It usually occurs in the 3rd to 4th decades. Cases are often self-limited and visual symptoms resolve by 4–8 weeks.
Etiology
The exact etiology is unknown, but several associations have been reported including HLA-B7 and HLA-DR2 genetic haplotypes. Approximately 33 % of patients have a preceding viral illness prior to symptom onset which may be associated with meningeal symptoms. APMPPE has also been described in cases of thyroiditis, erythema nodosum, Wegener’s granulomatosis, polyarteritis nodosa, nephritis, sarcoidosis, and CNS vasculitis. Other infectious associations include Lyme disease, mumps, and tuberculosis. Exudative retinal detachment, disk hyperemia, and episcleritis may occur in association [29]. Cerebral vasculitis has been associated with APMPPE and requires treatment with systemic corticosteroids [30].
Pathogenesis
It is believed to be due to inflammation at the level or the choriocapillaris leading to hypoperfusion and ischemia of the RPE and photoreceptors. In later stages, the inflammed choroid and retina is replaced with RPE atrophy and hyperpigmentation.
Symptoms
Patients present with rapid loss of vision or less frequently with photopsias and central and paracentral scotomas. The symptoms are usually bilateral but asymmetric. Visual acuity depends on the extent of foveal involvement and visual loss is transient.
Clinical features
Anterior segment is normal and there may be mild vitritis. Fundus examination shows multiple bilateral yellow-white placoid lesions at the level of RPE and choroid 1–2 disk diameters in size located in the postequatorial region (Fig. 18.3). The lesions gradually fade over the course of 1–2 weeks. New lesions may appear in the periphery up to 3 weeks following onset (radially or linearly). Older lesions are replaced with RPE atrophy or hyperpigmentation. Disk edema, retinal vasculitis, and venous occlusions may be present. Exudative retinal detachment, disk hyperemia, and episcleritis may occur [29]. Systemic associations have been described with APMPPE including meningoencephalitis, nephritis, and hearing loss. Fatal cerebral vasculitis has rarely been associated with APMPPE.
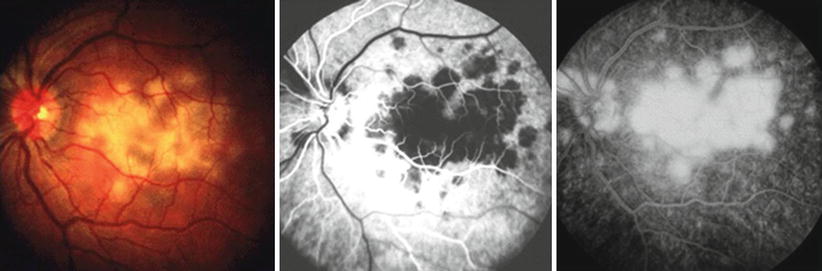
Fig. 18.3
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE)
Fluorescein angiogram (FA) shows early hypofluorescence (blockage) due to intracellular edema, leukocytic infiltration, and capillary nonperfusion. Hyperfluorescence in the late phases is due to leakage from choriocapillaries through damaged RPE cells.
Indocyanine green (ICG) angiogram shows early and late hypofluorescence corresponding to the placoid lesions.
Spectral-domain optical coherence tomography (SD-OCT) findings in APMPPE show that massive intraretinal edema may occur in the early stages [31].
Differential diagnosis
Multiple evanescent white dot syndrome [32] is more common in females.
Other infectious uveitis and lymphoma may present with placoid lesions as well and should be ruled out with appropriate tests if clinical suspicion is high.
Ampiginous choroiditis [1] is a separate disease entity due to its distinct clinical features. It is a disease with multiple relapses, which can be effectively controlled with a combination of immunosuppressive therapy.
Vogt-Koyanagi-Harada [33] syndrome can mimic APMPPE but FFA and if required a CSF study can help in differentiation.
Management
APMPEE is self-limiting and has a good prognosis. Visual acuity is diminished very profoundly in the early phases of the disease in some cases but recovers nearly completely to near normal levels. All patients with a new diagnosis of APMPPE should receive a full neurologic and systemic workup to evaluate for CNS vasculitis, sarcoidosis, and tuberculosis. Steroids are used if macular involvement is present as prompt use of systemic steroids resolves lesions rapidly. Cytotoxic therapy with cyclosporine and biologics [34] are used in the treatment of severe neurological disease if present. APMPPE resolves spontaneously in 2–12 weeks and does not usually require treatment. Intravitreal triamcinolone acetonide [35] improved macular anatomy, visual acuity, and macular sensitivity.
Complications
Some patients may have difficulty with reading due to persistent scotomas in the central field. Pigmentary alterations may remain after the resolution of the placoid lesions. Recurrences are rare as is choroidal neovascularization.
Birdshot Chorioretinopathy (BSCR) or Vitiliginous Choroiditis
BSCRC is a bilateral granulomatous, inflammatory disease associated with human leukocyte antigen (HLA)-29 antigen in 90 % of cases [36].
Clinical features
Presentation is in middle age with preponderance in women with decreased visual acuity, color vision, floaters, and nyctalopia. Anterior chamber is quiet but vitritis is typical. Oval, creamy lesions, ¼ to ½ disk diameter in size, are scattered in the posterior pole (Fig. 18.4). Fluorescein angiography shows hypofluorescence in the early phase and hyperfluorescence in the late phase. Indocyanine green angiography reveals well-delineated hypofluorescence choroidal spots in the midphase of the study which are far more numerous than those seen on either fluorescein angiography or clinically [37]. Optical coherence tomography (OCT) shows photoreceptor atrophy in several areas of both eyes with RPE degeneration beneath the areas of photoreceptor involvement [38]. Fundus autofluorescence [39] imaging shows hypoautofluorescence in areas of RPE atrophy that are more numerous and not uniformly correspondent with the birdshot lesions. Visual fields show progressive loss during the course by both Goldmann and automated (30-2) methods [40]. Electroretinography (ERG) is a useful tool to both diagnose and assess the response to therapy [41]. In specific, the 30-Hertz photopic ERG shows delayed implicit time and decreased signal amplitudes and this abnormality has been observed in 70% of patients at baseline. Apart from cone B wave implicit times, dim rod scotopic amplitudes are also significantly attenuated and decline continues with progression of the disease.
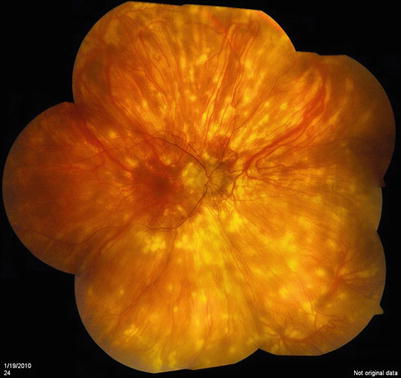
Fig. 18.4
Birdshot chorioretinopathy (BSCR)
Complications are retinal vasculitis, CME, and disk edema during active disease. Late complications include optic atrophy, epiretinal membrane (ERM), and CNVM.
Management is based on ERG. The ERG evolves into a negative pattern ERG, characterized by a decrease in b-wave amplitude. In advanced birdshot retinochoroidopathy, both a-wave and b-wave amplitudes are decreased, suggesting dysfunction of all retinal layers, including the photoreceptors.
Choroidal disease will readily respond to systemic inflammation suppressive therapy, as evidenced by normalization of ICGA findings. Though long-term corticosteroids have been tried, cyclosporine [42] in low doses of 2.5 mg is found to resolve lesions best. Other immunomodulatory therapies have been described with the use of mycophenolate mofetil [43] combined with cyclosporine, methotrexate [44], immunomodulatory agents [45], and biologics [46]. Prolonged treatment is required as there is progressive retinal dysfunction [47].
Differential diagnosis
Sarcoidosis, pars planitis, VKH syndrome, sympathetic ophthalmia, and POHS.
Multifocal Choroiditis with Panuveitis
The disease, typically bilateral, affects otherwise healthy individuals in their second to sixth decades of life [48]. Most patients are myopic females. Though it is a form of panuveitis, this is classified as a white dot syndrome due to the characteristic fundus appearance.
Etiology
The etiology of multifocal choroiditis is unknown; however, some propose that antigens in the retinal photoreceptors and retinal pigment epithelium become sensitized by an exogenous pathogen. There have been systemic associations of multifocal choroiditis including Epstein-Barr virus infection [49]. Multifocal choroiditis with panuveitis (MCP), PIC, and subretinal fibrosis and uveitis syndrome (SFU) represent a subset of white dot syndromes and are viewed as a single disease with a variable severity spectrum. Uncertainty also remains regarding the classification of multifocal choroiditis, punctate inner choroidopathy, and diffuse subretinal fibrosis as separate diseases or a spectrum of the same disease [50].
Symptoms
Patients typically present with decreased vision, photopsias, and enlarged blind spot [51].
Clinical features
Vitritis is present in all patients and may be associated with iridocyclitis. The classic lesions of multifocal choroiditis are 50–350 μm, punched-out chorioretinal scars with pigmented borders in the posterior pole. Acute lesions are typically yellow-white and located at the outer retina and choroid. They may number up to several hundred throughout the retina and they can occur in linear clusters or streak lesions as well. As these lesions age and become inactive, they become atrophic and punched-out. Over time, peripapillary scarring and atrophy may develop, and peripapillary and macular choroidal neovascularization may develop. Cystoid macular edema may be associated with acute lesions, while choroidal neovascularization may be associated with the juxtapapillary scars and deep macular scars [52].
Fluorescein angiography may highlight lesions not visible on clinical examination. Acute lesions show early hypofluorescence and late hyperfluorescence. Atrophic lesions appear hyperfluorescent in the early phases and fade in the late phases of the angiogram. Neovascular membranes are clearly demonstrated by FA [53].
Indocyanine green angiography may demonstrate multiple hypofluorescent spots compatible with active choroiditis around the optic disk.
SD-OCT shows drusen-like material between the RPE and Bruch’s membrane, vitreous cells, and localized choroidal hyperreflectivity [54].
Humphrey visual field often demonstrates an enlarged blind spot [55] and in some cases peripheral visual field loss that does not correspond to areas of acute choroiditis.
Complications
Cystoid macular edema and choroidal neovascularization cause vision loss. Retinal pigment epithelium metaplasia and fibrous scarring are rarer causes of vision loss. Secondary CNVM is very common in patients with MCP and is a frequent complication in up to 33 % of patients [56].
Management
The diagnosis is made by clinical examination but other entities must be ruled out with appropriate laboratory testing [57] and imaging studies [58]. Multifocal choroiditis has a waxing and waning course. These patients require long-term follow-up and visual prognosis is variable. Early in the course of the disease, systemic or periocular steroids are effective in controlling the disease. Late disease stages that include choroidal neovascularization and subretinal fibrosis require immunosuppressive agents for better control. Laser photocoagulation, photodynamic therapy, and anti-VEGF treatment are useful for CNVM. A comparison of bevacizumab versus photodynamic therapy for CNVM in MCP showed greater beneficial effects of visual acuity and central macular thickness in the bevacizumab group [59]. If there is extensive fibrosis that occurs with this syndrome, this may spill over into the development of progressive subretinal fibrosis and uveitis syndrome.
Differential Diagnosis
(a)
Presumed ocular histoplasmosis syndrome (POHS) where lesions are less numerous and smaller with no vitreous activity [60].
(b)
Infective multifocal chorioretinal lesions can present as a diagnostic and therapeutic dilemma. Laboratory workup is useful in determining an etiology; however, more invasive procedures, such as chorioretinal biopsy, may be necessary to guide treatment [61].
(c)
Birdshot retinochoroidopathy.
(d)
Sarcoidosis.
Punctate Inner Choroidopathy (PIC)
Punctate inner choroidopathy (PIC) is an idiopathic inflammatory disorder of the choroid which was first described by Watzke et al. in 1984 [62].
Etiology
Symptoms
The commonest reported initial symptoms are unilateral scotoma and blurred vision. The patient may be asymptomatic or present with scotoma. Acute loss of vision, however, could suggest choroidal neovascularization.
Clinical features
The disease is self- limited and is seen more frequently in myopic women with age range of 24–52 years [65]. Typical lesions are bilateral, multiple, small, well-defined, yellow-white, usually 100–200 μm diameter, and limited to the posterior pole. Lack of flare and inflammatory cells in the anterior chamber or vitreous cavity [66] is typical. The lack of vitreous inflammation is a hallmark of PIC and the presence of vitritis should suggest a different diagnosis [67]. They progress to atrophic scars, leaving a halo of depigmentation and are deeper and appear punched-out. Choroidal neovascular membranes occur in between 40 and 75 % of patients [68].
Fluorescein angiography (FA) shows that the active lesions are hyperfluorescent in the early phase with staining in the late phase. Evidence of complications such as leakage in the presence of cystoid macular edema (CME) and choroidal neovascular membrane (CNVM) is also seen. More lesions were seen on FA than on clinical examination. As the disease progresses, damage to the RPE occurs and FA demonstrates punctate RPE window defects.
Indocyanine green angiography shows areas of hypofluorescence corresponding to the choroiditis. Unusual abnormalities of the choroidal vasculature in PIC and the presence of larger choroidal vessels running through the hypofluorescent areas could imply that the vasculitic process is confined to smaller choroidal vessels and the choriocapillaris [69].
Optical coherence tomography
OCT shows attenuated signals of photoreceptor inner/outer segment areas corresponding to the visual field defect and increased choroidal thickness [70]. Spectral-domain OCT characterized a 5-stage evolution of PIC lesions as choroidal infiltration, formation of sub-RPE nodules, and then chorioretinal nodules, regression, and retinal herniation [71].
Fundus autofluorescence
Imaging of patients with MFC/PIC reveals extensive pathology beyond the clinically apparent spots. FAF findings highlight the areas of corresponding anatomic disruption of the photoreceptor–retinal pigment epithelium complex [72] and help to assess the resolution of the disease by the appearance of the RPE [73].
Electroretinogram (ERG) is typically normal.
Visual fields show enlargement of the blind spot in approximately 41 % of cases and central and paracentral scotoma. In many patients the blind spot extends toward the macula probably due to the peripapillary clustering of the inflammatory lesions.
Complications
CNVM and subretinal fibrosis. CNVM is the most common cause of visual loss in PIC. No differences were observed between eyes with typical and atypical choroidal lesions, supporting the notion that they represent a spectrum of a single disease, PIC [74].
Differential diagnosis
The multifocal dots in the posterior pole may resemble presumed ocular histoplasmosis, APMPPE, Behcet’s disease, Harada disease, leukemia, myopic degeneration, multiple evanescent white dot syndrome (MWEDS), pars planitis, sarcoidosis, sympathetic ophthalmia, serpiginous choroiditis, Vogt-Koyanagi-Harada disease, or Whipple’s disease.
Management
No treatment is advised for the majority of patients with PIC when there is no evidence of CNV as the visual prognosis is excellent.
Treatment if required may be as follows:
1.
Systemic corticosteroids.
2.
Multimodal approach: PDT combined with oral prednisolone (1 mg/kg body weight/day) which was started 5 days before PDT over a 12 month follow-up period and found a mean improvement in vision of 15 letter following a mean of 2 PDT treatments [77].
4.




Intravitreal dexamethasone implant [79].
Visual acuity improves with implants containing 0.7 or 0.35 mg of dexamethasone which releases the medication over a 6-month period.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
