Visual Dysfunction from Lesions of the Cerebral Cortex
Mathias Abegg
Jason J.S. Barton
The visual inputs to the primate visual cortex are functionally segregated. Information is relayed from the retina through the anterior visual pathway and optic radiations by at least two separate major pathways, the magnocellular (M) and parvocellular (P) pathways, which originate from different classes of retinal ganglion cells and project to separate layers of the lateral geniculate nucleus of the thalamus.1 The parvocellular pathway, which is relayed via layers 3 to 6 of the lateral geniculate body, is characterized by color opponency and slow-conducting axons that convey sustained signals.2,3 In contrast, the magnocellular pathway, relayed by layers 1 and 2, is characterized by large, fast-conducting axons conveying information about transient motion signals. These pathways terminate in separate divisions of layer 4C of the striate cortex.4
Beyond striate cortex, a large area of cerebral cortex is involved in visual processing (Figs. 22-1 and 22-2). As with the subcortical inputs to the striate cortex, the visual processing in extrastriate cortex also shows specialization. In a broad sense, this also involves two main parallel but interconnected streams, the ventral ‘what’ occipitotemporal stream and the dorsal ‘where’ occipitoparietal stream.5 These streams do not map directly in a one-to-one fashion with the subcortical M and P pathways,6 although M input may dominate in the dorsal pathway.7
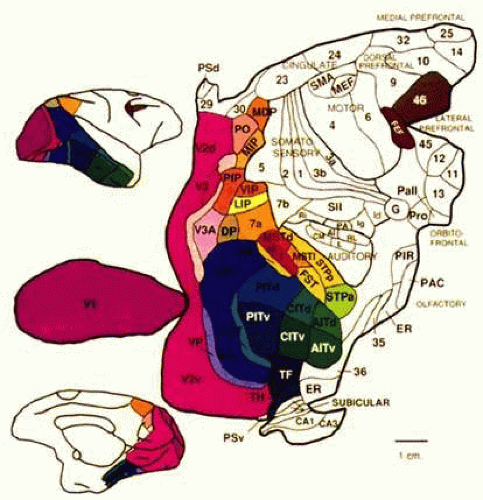 Figure 22-1. Visual cortex of the monkey. The monkey’s cortex is depicted as unfolded and splayed flat. Area V1 (Brodmann area 17) is the main recipient of parallel subcortical channels and a bottleneck for ascending inputs to maps of the visual fields located in the extrastriate cortex. Area V1 connects reciprocally with V2, V3, and surrounding regions in the early visual association cortex. It also connects directly and reciprocally with areas V5 (MT) and at least part of area V4. Area V4 receives a balance of M (magnocellular) and P (parvocellular) subcortical inputs; projects ventrally toward inferotemporal cortex (area IT); contains neurons modulated by attention, relevance, and perceptual context5,6 and may contribute to color and pattern processing.7,8 In contrast, V5 (MT) receives a predominance of M inputs and projects dorsally toward the parieto-occipital regions. The neural complex that includes area MT and surrounding regions9,10 is probably important for the processing of motion, attention, and related visuospatial processes.11 Human vision and its disorders can be explained in terms of a homologous organization. (Reproduced from Felleman DJ, Van Essen DC: Distributed hierarchical processing in the primate cerebral cortex. Cereb Cortex 1:1, 1991, with permission.) |
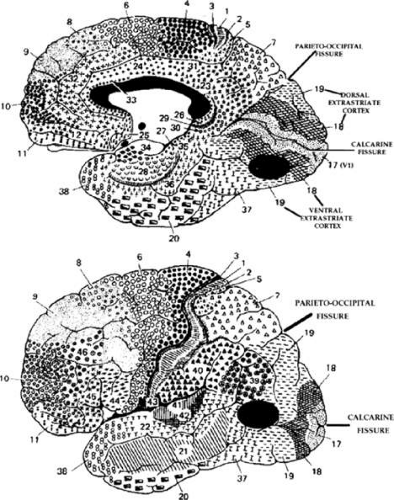 Figure 22-2. Human visual cortex. Brodmann (1909) divided the human cortex into several different cytoarchitectonic areas. He designated striate cortex as area 17 and showed it as symmetrically surrounding the calcarine fissure on the mesial surface of the hemispheres. Simian area V1 is located more laterally (Fig. 22-1). The foveal representation is located toward the occipital pole, whereas the peripheral visual field representations are located anteriorly, in the depths of the calcarine fissure. The parieto-occipital fissure is a fairly reliable anterior boundary for dorsal area 17. Extrastriate areas 18 and 19 concentrically surround area 17 and should correspond to the monkey’s areas V2 and V3. Top. Ventral 18 and 19 (including the fusiform and lingual gyri) and adjacent temporal lobe areas contribute to a “what” pathway, which should contain regions corresponding to simian areas V4 (dark oval area) and IT. Bottom. Dorsal 18 and 19 extend onto the lateral surface of the hemisphere and contribute to a parietal or “where” pathway, which may contain a human homologue of the monkey’s area MT (dark oval area) in portions of 19, 37, and 39. |
Information about color, luminance, and pattern recognition is processed mainly in the ventral “what” pathway. In humans it extends from below the calcarine fissure into the visual association cortex in the adjacent medial temporal lobe, which is thought to contain a human homologue of simian areas V4 and inferotemporal cortex (IT). Damage to this pathway is associated with impairments in recognition of objects (the agnosias), reading (the alexias), and color perception (cerebral achromatopsia). Information about motion perception and other visuospatial functions is thought to be carried by the dorsal “where” pathway. In humans it extends from the dorsal bank of the calcarine fissure into visual association cortices over the superior and lateral surfaces of the posterior hemisphere, which is thought to contain a human homologue of the monkey’s area middle temporal (MT) complex and parieto-occipital areas. Damage to this pathway affects control of visually guided eye and hand movements (as in Balint syndrome), and can impair motion perception (cerebral akinetopsia).
LESIONS OF THE STRIATE CORTEX
The primary visual area in the medial occipital lobe goes by several names: Brodmann area 17, visual area 1 or V1, calcarine cortex, and striate cortex. The exact position of the striate cortex varies between individuals. Whereas the parieto-occipital fissure forms a reasonably reliable anterior dorsal boundary, the posterior limit containing the macular representation is more variable, extending from the medial occipital surface to the first 1 or 2 cm of the posterior surface of the occipital lobe (Fig. 22-2).
The main vascular supply of striate cortex derives from the posterior cerebral artery. A parieto-occipital branch supplies the superior calcarine bank, a posterior temporal branch supplies its inferior bank, and a calcarine branch supplies the central region posteriorly. Individual variation exists, however.8 It is important to note that the occipital pole is at the junction (watershed zone) of the vascular territories of the posterior and middle cerebral arteries, and there is again marked variability as to which artery supplies the macula representation in striate cortex.8
The retinotopic arrangement in striate cortex has been known for nearly a century9,10 and confirmed with modern neuroimaging.11,12 The macula representation is posterior, at the occipital pole, and the far peripheral field is anterior, on the medial occipital surface. The superior bank of the calcarine fissure receives input from the contralateral inferior visual field, whereas the inferior bank contains the representation of the superior visual field. The most anterior part of the striate cortex represents the monocular temporal crescent, the region of temporal field in the contralateral eye that lies beyond the limits of the nasal field (60°) of the ipsilateral eye. As in most of the visual system, there are fewer neurons devoted to peripheral vision than to central vision: More than half of striate cortex is devoted to the central 10° (cortical magnification).11,12 The occipital cortex contains a mixture of monocular and binocular cells arranged in ocular dominance columns, but large separations between the inputs of the two eyes are not present.
VISUAL FIELD DEFECTS FROM STRIATE LESIONS
Complete destruction of the striate cortex causes complete visual loss in the contralateral visual hemifield. Because this involves not only peripheral vision but also the contralateral half of the foveal region, it is called a macula-splitting homonymous hemianopia. This may occur with posterior cerebral artery ischemia in a patient whose entire striate cortex is supplied by that artery. Macula-splitting hemianopias can occur with complete lesions anywhere along the retrochiasmal visual pathways and thus lack localizing value.
In cases of incomplete homonymous hemianopia, it was recognized early that occipital lesions lead to highly congruent visual field defects, meaning that the defects are almost identical in the two eyes, whereas more anterior lesions in the optic radiation or optic tract result in incongruent field losses.13 A study of more than 500 patients with incomplete homonymous hemianopia confirmed this observation but showed that the rule is a relative one. Occipital lesions result in a congruous defect in 84% and in an incongruous defect in 16%, whereas optic tract lesions cause congruous defects in 50% of cases.14 Congruency is also more likely after a stroke than when caused by trauma or a tumor.14,15
With posterior cerebral infarcts, a macula-sparing hemianopia (Fig. 22-3) occurs in patients with adequate collateral circulation of the macula region (occipital pole) from the middle cerebral artery.8 Traditionally, macula-sparing has been considered almost diagnostic of a striate lesion. However, a review of 904 cases found that only 51% of all macular-sparing homonymous hemianopias were a result of occipital lesions, with the rest associated with lesions of the optic radiations or tract.16
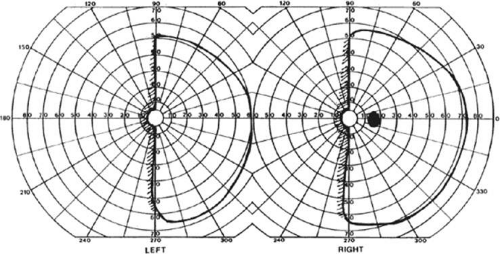 Figure 22-3. Macular-sparing homonymous hemianopia. Visual field by Goldmann perimetry (isopter to V4e target) in a 49-year-old woman with a right medial occipital infarct sparing the occipital pole. |
The upper and lower banks of the striate cortex can also be involved separately. Ischemia can do this because the banks have separate blood supplies. Upper calcarine bank infarcts cause homonymous contralateral inferior quadrantanopia (Fig. 22-4), and lower calcarine bank infarcts cause superior quadrantanopia. With some exceptions,17,18 most quadrantic defects do not align at the horizontal meridian, because the lower field transitions smoothly into the upper field in the depths of the calcarine fissure, without any anatomic separation. Quadrantanopias are more common with striate damage than with optic radiation lesions.16,19
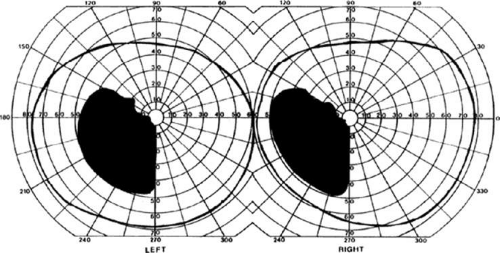 Figure 22-4. Left inferior quadrantanopia sparing the macula but not respecting the horizontal meridian. Visual field by Goldmann perimetry (isopter to V4e target) in a 50-year-old woman with a striate infarct involving mainly the superior bank of the calcarine fissure. |
Selective lesions can also occur along the anterior-posterior extent of striate cortex. A lesion of the occipital pole alone causes homonymous central hemiscotomata20,21 (Fig. 22-5). This may occur with watershed infarcts during systemic hypoperfusion. Slightly more anterior lesions in the middle zone of striate cortex cause homonymous peripheral scotomata (Fig. 22-6). The homonymous nature of these defects and their restriction to one hemifield differentiate these from ocular causes of central or paracentral visual loss. Lesions with such small field defects can be missed on CT.22 MR with coronal sections through the occipital lobes should be performed, although even this may miss small lesions, particularly at the occipital pole.
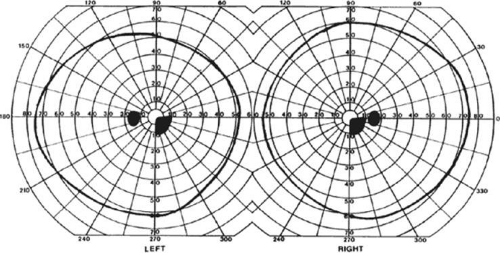 Figure 22-5. Homonymous right inferior central scotomata. Visual field by Goldmann perimetry (isopter to V4e target) in a 38-year-old man with sudden complaints of difficulty reading. MRI showed an infarct of the left occipital pole. |
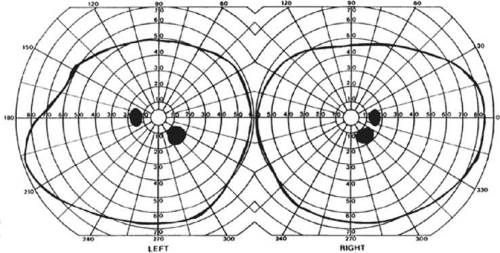 Figure 22-6. Homonymous right inferior paracentral scotomata. Visual field by Goldmann perimetry (isopter to V4e target) in a 14-year-old girl with perinatal occipital infarction. MRI showed a lesion of the midportion of the left superior calcarine bank, sparing both the occipital pole and the anterior striate cortex. |
A near-complete lesion that spares only the most anterior portion of V1 causes a hemianopia with sparing of the monocular temporal crescent (Fig. 22-7). The hemianopia involves the whole nasal hemifield of the ipsilateral eye, but the temporal hemianopia of the contralateral eye spares a crescent-shaped island of vision in the far periphery.23 This is the region of the visual field that is represented in the temporal field of one eye but not the nasal field of the other. The initial sense of incongruity may raise suspicions of an optic tract lesion; however, the absence of optic atrophy and relative afferent pupillary defect (RAPD), the high congruity of the homonymous defect inside 60°, and the location of the crescent outside 60° eccentricity, indicate that the lesion is probably cerebral. Sparing of the temporal crescent can occur with lesions of either the striate cortex or optic radiation.24 The converse defect, a monocular temporal crescentic scotoma, can occur with a lesion of cortex just behind the splenium of the corpus callosum, along the parieto-occipital sulcus.25
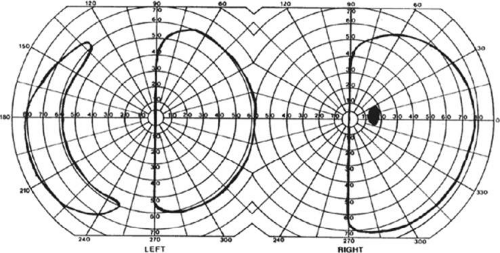 Figure 22-7. Macular-splitting hemianopia with sparing of the monocular temporal crescent. Visual field by Goldmann perimetry (isopter to V4e target) in a 66-year-old man with a right medial occipital infarct. |
Additional neurologic defects may be helpful in determining the site of the lesion causing hemianopia. Hemisensory or hemiparetic defects suggest a lesion to the parietal lobe rather than the occipital lobe, although these can also occur with posterior cerebral arterial infarcts if there is associated thalamic damage. Damage to medial occipitotemporal regions causes amnesia, prosopagnosia, and color perception defects. A syndrome of agitated delirium and hemianopia occurs with lesions of the medial occipital lobe, the parahippocampus, and hippocampus.26,27,28 Brainstem signs from vertebrobasilar ischemia can accompany posterior cerebral artery strokes: These include impaired level of consciousness, third nerve palsy, dysarthria, and hemiplegia.29
The most frequent cause for a homonymous hemianopia is stroke (70%), from either cerebral infarction (59%) or hemorrhage (11%).16 Trauma is the cause in 14%, while a neoplasm is found in 11%. Infrequent causes include iatrogenic neurosurgical trauma (2%), demyelination (1%), and various metabolic or degenerative conditions in <1%, including Alzheimer disease, prion disease, MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes), epilepsy, and corticobasal ganglionic degeneration (Fig. 22-8).
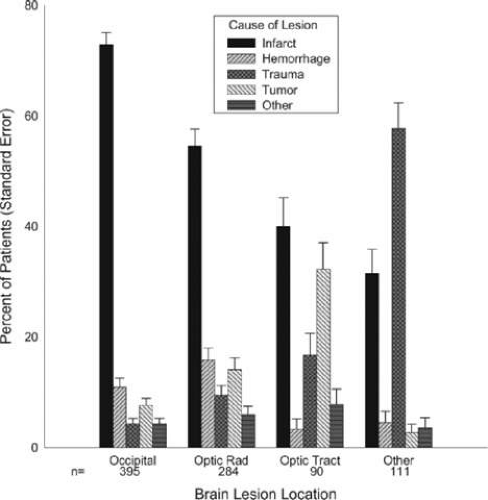 Figure 22-8. Etiology of field defects. (Reproduced from Zhan X, Kedar S, Lynn MJ et al: Homonymous hemianopias: Clinical-anatomic correlations in 904 cases. Neurology 66(6):906–910, 2006, with permission from Wolters Kluwer Health [Lippincott Illustration].) |
Bilateral hemifield defects are unusual but do occur. Bilateral incomplete hemianopia is distinguished from bilateral optic nerve or ocular disease by the congruity of the visual fields and step defects along the vertical meridian, which indicate the hemifield nature of the visual loss.30 Such steps are important to seek with a skilled perimetrist, but even so they can be difficult to demonstrate with bilateral central hemiscotomata from occipital pole lesions.31 Bilateral quadrantanopias can occur,17,18 often in patients with prosopagnosia and achromatopsia, for example, and may mimic the altitudinal defects of optic neuropathy, except that they are binocular. The most common cause of bilateral hemifield defects is posterior circulation ischemia.30 This can affect both striate cortices either simultaneously or sequentially,30 as the right and left posterior cerebral arteries have a common origin from the basilar artery. Twenty-two percent of patients with a unilateral occipital infarct will develop bilateral infarction over 3 years.32 Other causes of bilateral hemifield defects include focal midline lesions such as tumors or traumatic injury, since the right and left striate cortices face each other on the medial occipital surface.
CEREBRAL BLINDNESS
Sometimes cerebral lesions cause complete loss of vision. This is distinguished from ocular disease by both normal pupillary light responses and normal funduscopic examination, findings that may lead to an erroneous diagnosis of factitious visual loss. Associated signs of parietal or temporal damage may point to a cerebral cause, but these are not always present. Visual evoked potentials are of limited diagnostic value. They can be altered voluntarily by subjects without visual loss34 and can be normal in patients with striate lesions.35,36 They cannot differentiate between blind and seeing children with neurologic disease,37 and the results do not predict visual outcome.38,39 Absent evoked responses are rare and may only occur early in the course.37 Absent alpha rhythm on electroencephalography40,41 may be a more sensitive diagnostic sign.38 CT scans can be normal, but modern magnetic resonance imaging (MRI) with coronal images through the occipital lobe should reveal most striate or optic radiation lesions with complete and persistent visual loss. Single photon emission computed tomography (SPECT) scans may reveal bilateral functional defects in cases with unilateral MRI lesions.42
Cerebral blindness can be persistent or transient. The most frequent cause of persistent cerebral blindness is infarction.38 In addition to the common causes of emboli or atherosclerosis, it has also been reported with vertebrobasilar arteritis,43 subclavian steal,44,45 and hypotension from antihypertensive medication.46,47 Cerebral blindness can complicate cardiac surgery, because of hypotension or emboli.38
Transient cerebral blindness can last from hours to days, often followed by full recovery. It can occur with or following a seizure,48,49,50 with metabolic insults,51,52,53 hypertensive encephalopathy,54 pre-eclampsia,55 hydrocephalus,56 trauma,40,57 and cortical venous thrombosis.58 Toxic causes include chemotherapeutic agents,59,60,61,62,63 immunosuppressants like tacrolimus64 and cyclosporin,65 and iodinated contrast agents used in angiography,38,66,67 where a CT scan with contrast can show disruption of the blood–brain barrier in the occipital lobes66,67 as early as 1 hour after angiography.68
The pathogenesis of transient cerebral blindness varies with the cause. Vasogenic edema is probably the chief factor in the posterior reversible encephalopathy syndrome (PRES) that results from seizures, chemotherapy, and hypertensive encephalopathy.69 Angiographic contrast agents may cause breakdown of the blood–brain barrier under osmolar stress and subsequent neurotoxic effects,68 providing a rationale for treating with dexamethasone and mannitol in this context. MRI using diffusion-weighted imaging and apparent diffusion coefficient images can distinguish vasospasm from stroke and help determine the prognosis for recovery of visual loss.69
As well as vasogenic edema, vasospasm may play a role in head trauma,70 eclampsia,55,71 methamphetamine abuse,72 and meningitis, which may also induce vasculitis.73,74
Transient cerebral blindness occurs in about 1% to 15% of patients with pre-eclampsia or eclampsia.55,71,75 Fluctuations in blood pressure with peripartum blood loss may contribute.76 The pathogenesis is analogous to that of hypertensive encephalopathy, with petechial hemorrhages and focal edema from impaired vascular autoregulation, and ischemia from vasospasm.55,77 CT scans may be normal or show bilateral occipital lucencies.55,78 MRI is more sensitive, showing T2 hyperintensities and T1 hypointensities, similar to hypertensive encephalopathy.77,79 Management is that of eclampsia, with magnesium sulfate, fluid restriction, and blood pressure control. The prognosis is good, with virtually all women regaining vision within a few hours to a week, rarely as long as 3 weeks.55 The differential diagnosis of eclamptic visual loss includes cerebral venous thrombosis80 and systemic hypoperfusion associated with pulmonary emboli.81 Antiphospholipid antibodies may be associated with infarction or venous thrombosis.82
Pediatric cerebral blindness is not uncommon. Children may not complain of visual loss but present with agitation, disorientation, and unsteadiness.41,70,83 During tests of vision they may not respond or may confabulate answers.83,84 Further observation shows that they do not fix, follow, or make saccades to objects, or do not show optokinetic responses.37,41,84 Major causes include head trauma, bacterial meningitis,73,74,84,85 and hypoxia from cardiac or respiratory arrest.84 Other causes include encephalitis, metabolic53,84,86 and toxic72 conditions. Seizures are common, and mental retardation is a frequent long-term complication among patients with meningitis.85
Posttraumatic transient cerebral blindness occurs more in children and young adults following apparent minor head injury,41,87,88,89 and in about 1% of closed head injuries.40,57,90 The impact is often in the parieto-occipital region.40,41,89,91 Headache, confusion, anxiety, nausea, and vomiting are frequent,89 as is loss of consciousness at the time of injury.57 Blindness may occur immediately or after a few minutes,41,57,89,91 sometimes after a few hours in adolescents.57 Vision recovers within 24 hours89,91 although the course in adults is more variable, with occasional residual deficits.57 CT of the head often is normal,83,89,91 but electroencephalography (EEG) shows bilateral occipital slowing.41 Protracted recovery, especially in children, may indicate cerebral contusions91 or occipital lobe infarction from tentorial herniation.92
Cerebral blindness can lasts hours, days, or weeks, recover fully, evolve into a hemianopia or other field defects, or be permanent.39,84,85 If there is recovery, it usually occurs within 5 months.39 Recovery is more likely in children than in adults.84 The prognosis is better with hypotension during cardiac surgery than with other causes.39 Preterm infants suffering hypoxia have a poor outcome, as they tend to have periventricular leukomalacia: Optic radiation damage on their CT or MR imaging is a poor sign.93 Normal MR imaging in most other settings is a good prognostic sign,39,93 and significant recovery can occur if hypoxic damage is limited to the visual cortex.93 Spike-and-wave discharges on EEG are associated with multiple handicaps and poor visual recovery.39
ANTON SYNDROME
About 10% of patients with cerebral blindness are not aware of their deficit and insist that they can see.30,38 This denial syndrome may be considered a form of anosognosia (denial of acquired impairment). Patients confabulate responses and when confronted with their errors, they often make excuses such as “I don’t have my glasses.” Anton syndrome is usually associated with bilateral cerebral infarcts in the primary visual areas of the calcarine cortex and visual association areas. Symptoms have been attributed to right hemispheric dysfunction or disconnections between the thalamus and the right parietal lobe.94
Denial of blindness on its own is not pathognomonic of cerebral blindness, as it can also occur in patients with ocular or optic nerve disease when there is concurrent dementia or a confusional state. However, altered mental status is not a necessary accompaniment of denial of blindness with lesions of the visual cortex. Thus, Anton syndrome should be more specifically defined as the denial of blindness in the absence of dementia or delirium. However, even cases meeting this more restrictive definition do not always have a lesion of the visual cortex, since such a presentation can also result from unusual combinations of bilateral optic neuropathy and bilateral frontal lobe disease.95
ABNORMALITIES IN THE REMAINING VISUAL FIELD
Some patients with hemifield visual defects curiously have complaints about the vision in their remaining good field, such as visual fatigue or blurring, especially in tasks with high attentional demands like reading, finding a face in a crowd, or driving a motor vehicle. There is evidence that unilateral lesions produce not only contralateral scotomata but also deficits in the remaining field, both ipsilateral and contralateral to the lesion. There are spatial and temporal contrast sensitivity deficits in the supposedly normal ipsilateral hemifield,96 reduced sensitivity and increased response times to signals in both hemifields, and reduction in the useful field of view.97 Saccadic response times to targets in the ipsilateral hemifield are also reduced.98
The origins of these subtle bilateral effects of unilateral lesions are unclear. One hypothesis is that white matter damage disrupts projections from V1 to other visual areas, callosal connections between the two hemispheres, and/or feedback projections from higher visual areas. A lesion in one area can thus affect the functioning of another more distant area (diaschisis). Disrupted connections can impair the synthesis of information, producing long-range disturbances in both hemifields, outside the confines of the limited contralateral scotoma. Damage to extrastriate regions may have similar bilateral long-range effects, as shown in monkeys with lesions of area V4.99 Thus associated extrastriate damage may contribute to impaired processing efficiency in the ipsilateral field of patients with hemianopia.97
REHABILITATION OF HOMONYMOUS DEFECTS
Some spontaneous improvement of homonymous hemianopia is seen in at least 50% of patients first seen within 1 month of injury.100 A scotoma that persists beyond 6 months is almost always permanent. The larger and closer to fixation the scotoma, the worse the effects on daily activities such as reading and driving.101,102 Rehabilitation attempts may be classified into the following categories: optical adaptations, strategic adaptations of eye movements, or attempts to restore visual field (reviewed in Schofield and Leff103).
Optical Devices to Promote Adaptation
This consists of moving the intact visual field of one eye into the blind field of the other eye. This may be achieved with prisms that may be monocular or binocular, covering only part of the lens or the entire lens. Monocular sector prisms provide field expansion only when the gaze is directed into the prism. The central diplopia accompanying the field expansion may be disorienting to the patient.104 In 2000, Peli105 described high-power press-on prism segments (40 prism diopters), that are placed across the whole width of the spectacle lens above and below the pupil area on the side of the field loss. Visual field expansion of about 20°, which is effective at all lateral positions of gaze, is provided via peripheral diplopia. In a study involving 32 patients, 74% continued to wear the prism glasses at 6 weeks, and at 8 weeks 47% were still wearing the prisms for a median duration of 8 hours per day, indicating that the use of gained peripheral vision outweighs the disturbing double vision in some patients.106
Strategic Adaptation of Eye Movements
Visual search training involves hemianopic patients practicing identifying objects in their blind and seeing hemifields. Training improved detection of and reaction to visual stimuli without restitution of the visual field defect. The improvement is achieved via increased efficiency of exploratory eye movements and parallels improvement in daily living skills.107 Such strategic adaptation also occurs naturally without training as long as subjects do not have additional hemineglect.108,109
Strategic adaptation of eye movements has also been studied in hemianopic dyslexia. Alongside the loss of driving privileges, impaired reading is the most significant functional impairment of hemianopia. Patients with right hemianopia produce many more saccades than normal subjects when reading a line of text, as the hemianopia deprives them of visual information about the shape and location of ensuing words that normal readers use to guide their reading saccades. Reading speed is improved when patients learn to make larger and more efficient saccades into their blind hemifields.110 Exposing patients to small-field optokinetic stimulation as a therapy further enhances this process.111
Attempts at Restoring Vision
The above methods do not change the region of visual loss, but help patients cope with them. However, several studies have suggested that training can actually reduce the region of blindness in hemianopia in both adults112,113,114,115 and children.116 The validity of some of these results has been questioned, as well as their interpretation, as to whether there was true recovery from neural plasticity, or if the results could be explained by shifts in the response bias of patients or their fixation patterns. For example, perimetric evaluation with scanning laser ophthalmoscopy, which allows precise fixation control, failed to confirm any improvement of the field, suggesting that the reported benefits may be attributable to fixation shift.117
In contrast to vision restoration therapy, which aims to expand the visual field from the borders of the intact field, another group has claimed to demonstrate restitution of function deep within the damaged visual field,118 possibly by utilizing extrastriate pathways involved in blindsight.119 Enhancement of these alternative routes by training may lead to increased performance and increased awareness.
EXTRAGENICULATE VISUAL PATHWAYS
The classical route for conscious vision involves retinal ganglion cells projecting to the lateral geniculate nucleus of the thalamus and from there to the primary visual cortex. However, some fibers leave the classical visual pathway before it reaches V1 (Fig. 22-9). Some ganglion cells project to the pretectal nuclei of the midbrain to mediate the pupillary light response. Others may contribute to ocular motor functions via projections to the superior colliculus, accessory optic system, nucleus of the optic tract, olivary nucleus, and ventral lateral geniculate nucleus.
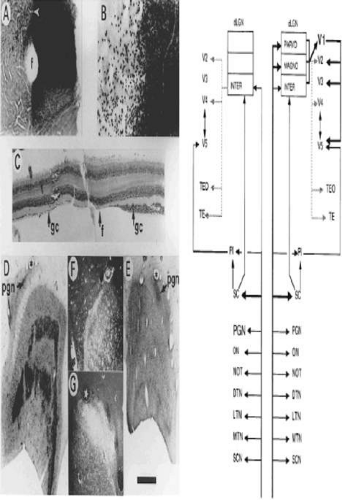 Figure 22-9. Extrastriate visual pathway. A. Photomicrograph of flat-mounted retina of a macaque monkey in which the upper half of the left optic nerve was labeled with horseradish peroxidase 4 years after removing the left striate cortex at the age of 5.5 years. The normal nasal hemiretina is densely stained, in comparison with the retrogradely transneuronally degenerated temporal hemiretina to the left of the fovea (f). B. Higher-power photomicrograph centered on the white arrowhead, also shown in A. The loss of ganglion cells is substantial. C. Horizontal section (10 μm) in the vicinity of the fovea, through the other eye of the same monkey. The degenerated ganglion cell layer of the nasal hemiretina, normally slightly thicker than the temporal hemiretina, has been reduced to a monolayer. The section had cracked to one side of the fovea and has been realigned. D and E. Sections of the normal and degenerated dLGN of a macaque monkey in which the striate cortex was removed unilaterally at the age of 18 months and the animal was perfused 22 months later, following an injection of [3H]proline into the eye contralateral to the ablation. Dark bands, prominent in D but faint in E, show by autoradiography, labeling of magnocellular and parvocellular layers. The pregeniculate nucleus (PGN) is both relatively and absolutely larger on the degenerated side. F and G. Dark-field photomicrographs of the PGN showing the more prominent input to the degenerated side, some but not all of which is attributable to the normally greater labeled contralateral input. The star and asterisk in D and F mark a corresponding capillary, as they do in E and B. The scale bar represents 1 mm for A, D, and E; 250 μm for B and C; and 625 μm for F and G. The schematic diagram shows known inputs from the eye to retinorecipient nuclei and some of the subsequent forward projections. The right half of the figure shows the normal condition, the left half the effect of removing the striate cortex (V1). The cortical pathways exclude further projections from V2, V3, V4, V5, and so on. For example, parts of the inferior pulvinar also project to V2, V3, and V4. Following removal of striate cortex, the PGN expands and the ON contracts, indicated by the size of the lettering. With respect to geniculocortical pathways, the dotted lines indicate relatively sparse projections. SCN 5, suprachiasmatic nucleus; MTN, LTN, DTN 5, medial, lateral, and dorsal terminal accessory optic nuclei; NOT 5, nucleus of optic tract; ON 5, olivary nucleus; PGN 5, pregeniculate nucleus; SC 5, superior colliculus; PI 5, inferior pulvinar. (Reproduced from Stoerig P. Cowey A: Blindsight in man and monkey. Brain 120(Pt 3):535–559, 1997, with permission from Oxford University Press.) |
In lower vertebrates a specific subset of ganglion cells contain a photosensitive pigment, melanopsin. Although they receive most of their input from photoreceptors, these melanopsin-containing ganglion cells can generate a pupillary light response even in the complete absence of classic photopigments. In mice and macaques, it is known that these cells also project to the suprachiasmatic nucleus and the intergeniculate leaflet, areas involved in regulating circadian rhythm. In the macaque some fibres from melanopsin-containing cells also project to the lateral geniculate nucleus and may transmit color and luminance information.120
BLINDSIGHT AND RESIDUAL VISION
Patients with visual loss resulting from lesions of the striate cortex or optic radiations may have some remnant visual function in their blind field (Table 22-1). Some of these patients retain some awareness of visual stimuli, and hence have residual vision,121,122,123 which may be difficult to distinguish from a severe relative visual field defect. Blindsight refers to a condition in which patients with visual field defects can perform at greater-than-chance levels on visual discrimination tasks, despite being unaware of the visual stimuli.124,125,126 It is unclear whether the pathophysiology of blindsight differs from that of residual vision—sometimes referred to as type I and type II blindsight, respectively.127 The phenomenologic distinction between the two is not necessarily sharp. Rather than a dichotomy between the presence and absence of awareness, the degree of consciousness in perception can vary along a spectrum.128,129 Also, stimulus parameters can be manipulated so that a patient shows residual vision under some conditions and blindsight under others.130,131
A recent taxonomy grouped blindsight phenomena into three clusters, based on purported anatomic substrates.132 Action blindsight includes manual and saccadic localization; attention blindsight includes residual functions such as covert orienting, inhibition of return and motion perception; and agnosopsia includes abilities to report on properties of stimuli such as color, form, and semantic content. Experimentally, blindsight studies can also be divided into those that use direct techniques, which require a response to the blind stimulus, such as detection, forced-choice discrimination, or localization, or indirect techniques, in which the experiment determines if responses to seen stimuli are influenced by stimuli in the blind field.
TABLE 22-1. Residual Visual Abilities Shown in Various Blindsight Studies | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Action Blindsight
Some studies find that saccades, reaching, and pointing to locations within hemianopic fields are weakly correlated with target position, mainly for a limited range of paracentral locations.123,124,128,133,134,135,136,137 Other studies have failed to find saccadic localization.98,138,139 An indirect approach showed that saccadic trajectories to a seen target deviated when distractors were present in the blind hemifield in some hemianopic subjects.140
Stimulus motion can modulate action blindsight. Target localization was easier with moving rather than stationary targets in some reports141,142 but not another.143 This is reminiscent of the Riddoch phenomenon,144 in which movement is appreciated before static targets in recovering hemianopic defects.144 Motion information might also guide eye movements. Recovery of optokinetic responses was reported in one patient with cortical blindness145 but not in two others137,146 nor in three hemianopic patients.147 Pursuit and saccadic responses to motion were not found in patients with medial occipital lesions sparing the lateral human motion area.98
Attention Blindsight
Studies show some perception of speed and direction of rapid bright spots in blindsight.121,123,151 With larger stimuli such as optokinetic gratings, some patients can discriminate motion direction147 or experience an illusion of self-motion.152 An indirect strategy showed that perception of optic flow within the intact hemifield could be enhanced by optic flow in the blind hemifield.153 Some of these motion abilities may not reflect real perception of velocity, but rather from the processing of spatial position or flicker: Studies with random dot stimuli that minimize these confounds found no residual motion perception in 13 patients.154,155
Spatial summation occurs when two simultaneous stimuli generate faster responses than a single stimulus; temporal summation is the decrease in reaction time when a stimulus is preceded by another. Evidence for summation between seeing and blind fields has been inconsistent and found in only a few subjects.136,153,156,157 A distractor effect, in which targets in the blind field slow response times to seen stimuli, has been shown for saccades but not manual reaction times.158 This finding could not be replicated in another study.159 Inhibition of return is a normal phenomenon in which a stimulus delays the detection of a target appearing in the same location a short time later. This phenomenon was generated in the blind hemifield of one patient.160
Agnosopsia
Early studies found no evidence of chromatic perception,123,135 but later work showed detection of colored targets161 and evidence of color-opponent interactions in the spectral sensitivity curves.162 Colour effects have also been studies using the pupil. While the standard pupillary light reflex that is clinically measured is a response to changes in luminance, there are also pupillary responses to gratings and isoluminant colors. Pupillary responses to such stimuli in the blind hemifield have been shown in one patient.163,164 In another study, color stimuli caused afterimages that evoked pupillary responses in normal subjects; in the blind hemifield of two patients, there were ‘after’ pupillary responses without any conscious afterimage.165
For form perception, studies of temporal and spatial contrast sensitivity have yielded mixed results, even when done on the same patient (subject GY).96,166,167 An early report claimed that one patient could discriminate large X and O forms, perhaps through orientation perception.126,135 However, form discrimination was not found in seven other patients,123,134,168 nor orientation discrimination in two others.148,169
Word perception can be influenced in blindsight. The choice of meaning of an ambiguous word (i.e., ‘light’) in the seeing field can be influenced by a word in the blind field (i.e., ‘dark’ vs. ‘heavy’).128 Completion effects have been reported for form perception. Two patients had completion effects for afterimages or with illusory contours such as the Kanizsa triangle.128 In an interference task, reaction times to seen letters and colors were prolonged by flankers different from the target, even when these were located in the blind field, in a patient with an occipital lesion.170
A direct pathway between the pulvinar and the amygdala may mediate reactions to frightening stimuli.171 One patient could process fearful and angry faces in his blind hemifield,172 and this may correlate with activity in his amygdala.173 Another study of a cortically blind patient showed that associating a visual stimulus with a painful shock can potentiate startle reflexes to that stimulus, despite the lack of conscious perception.174
Explanations of Blindsight
Blindsight studies require careful exclusion of artifacts as alternative explanations. Four issues need consideration. First, poor control of fixation may allow the subject to place the target in the seeing hemifield. Eye position monitors can help exclude this, as long as both head and eye position are controlled or detected.175 Second, light scatter may diffuse from blind-field stimuli into the seeing field and mimic blindsight.98,176,177,178,179 To counter this, some flood the seeing field with light. Attempts to control for scatter include testing the physiologic blind spot,126 and also testing control patients with pregeniculate lesions133,134,141,170 or hemispherectomies.147,179 Third, careful perimetry may reveal that the blind field has surviving islands of vision that account for the residual perception.180,181,182,183 Fourth, blindsight may simply represent a “criterion shift.” Subjects may reply more liberally when asked to choose between alternatives than when asked if they see something: This has been shown for normal subjects.176,184,185 Signal detection analysis has been used to rebut this criticism,161,186,187 although it has also suggested that changes in criterion bias may indeed explain some blindsight motion perception results.188 Relevant to these last two points are ongoing debates about whether blindsight resembles degraded normal vision or shows qualitative differences.129,189,190
Several different anatomic pathways are invoked to explain blindsight. Action blindsight such as spatial localization may involve an extrageniculate pathway from retina to superior colliculus,133,191 with possible further involvement of posterior parietal cortex. It is claimed that better blindsight in the temporal than nasal hemifield of patients is a signature of mediation by the superior colliculus.158,192 Residual visual abilities with achromatic but not blue-yellow stimuli are also cited as a signature of mediation by the superior colliculus, since S-cone inputs may not be represented in this structure.193,194 Attention blindsight may also be mediated via the superior colliculus, with additional relay to the extrastriate visual cortex such as area MT via the pulvinar. Since the superior colliculus lacks color opponency, chromatic blindsight161,162,170,195,196,197 may indicate yet another relay, perhaps direct projections from surviving LGN neurons, possibly in the intralaminar (koniocellular) layer, to the extrastriate cortex.198
The monkey evidence most strongly supports the retino-tecto-pulvinar relay to extrastriate regions, particularly those involved in motion perception. Lesions of V1 do not abolish responses in V5199,200,201 or V3A202 unless accompanied by lesions of the superior colliculus.199,203 On the other hand, one study using optical imaging found that deactivation of V1 with muscimol abolished activity in V5.204 Stabilization of an early direct projection from LGN to V5, which normally regresses with development, may occur in infant cats with striate lesions,205 but evidence of this is lacking in infant monkeys.206
Physiologic techniques in humans have also provided some support for these hypothesized pathways. In normal subjects, evoked potentials207 and some transcranial magnetic stimulation studies208 (but not others209) suggest that visual motion signals may arrive in V5 before and independent of V1. Positive emission tomography (PET) scans,122 magnetoencephalography,210 and evoked responses211 have shown residual activation of V5 by rapid but not slow-moving stimuli in the blind hemifields of one patient. An intriguing functional MRI study of patient GY (mentioned earlier) that varied stimuli to obtain responses with and without awareness showed that residual vision was associated with activation of extrastriate visual areas and dorsolateral prefrontal cortex, and blindsight with activation in superior colliculus and medial prefrontal cortex.130
Theories of blindsight must also account for its variability. Recent large series, representing 46 patients in total, suggest that blindsight is rare.154,181,182 One important factor may be the extent of additional damage to the optic radiations and extrastriate cortex. However, correlations between blindsight abilities and lesion anatomy have proven elusive.98,138,154,156,212 A requirement for very focal striate damage is also difficult to distinguish from a need for partial striate damage,153 pointing back to a potential artifactual explanation. Another important factor may be the timing of the lesion. Blindsight may require neural plasticity. If so, age at onset, time since lesion, and possibly training may be important.125 Infants or children may be more likely to develop blindsight or residual vision in both nonhuman primates and man.123,141,205,213 Some reports imply that up to half of children with cerebral blindness may have some residual vision for moving objects.214 Not all studies find that age matters, however.215
The efficacy of training in eliciting blindsight is controversial. Although some deny that training helps,123,175 others claim that practice can improve saccadic or manual localization.142,212,216,217,218 A study that trained nine hemianopic subjects with various forced-choice localization and form discrimination tasks found improvement after 5 to 6 months.219 Training of motion perception may improve performance in the retinotopic region trained.220 There are even claims that such training may expand visual fields.219,221,222
LESIONS OF EXTRASTRIATE CORTEX
Area 17 or V1 is concentrically surrounded by extrastriate areas 18 and 19. These areas likely correspond to the monkey’s area V2 and V3. Areas 18 and 19 are mostly medial: Their ventromedial aspects include portions of the fusiform and lingual gyri and may correspond to another visual area of the monkey, area V4. Like V1, the ventromedial parts of areas 18 and 19 are supplied by branches of the posterior cerebral artery; however, areas 18 and 19 also extend onto the lateral surface of the occipital lobe. Several studies suggest that a human homologue of the monkey’s area MT (V5) complex is present on the lateral surface of the brain, in a region where the occipital, temporal, and parietal lobes converge. This would include portions of Brodmann areas 19, 37, and 39. Like the occipital pole, the blood supply of these lateral regions falls in the watershed zone between the middle and posterior cerebral arteries.
Given our knowledge of parallel visual processing in the extrastriate cortex of monkeys, we can create two broad categories of visual dysfunction from human lesions: those affecting components of the ventral pathway, which disrupt form and color perception; and those affecting components of the dorsal pathway, which disrupt motion and visuospatial perception.
THE VENTRAL “WHERE” OCCIPITOTEMPORAL PATHWAY
DISORDERS OF COLOR PROCESSING
Cerebral Dyschromatopsia/Achromatopsia
Cerebral (central) achromatopsia is complete loss of color perception, whereas cerebral dyschromatopsia indicates some residual color perception, as is most often the case. Both are rare. Hemiachromatopsia is color loss limited to the contralateral hemifield223,224 and may be more common but under-recognized.
Achromatopsic patients are generally symptomatic, complaining that the world appears in shades of grey.225,226,227,228 Some also report that the world appears less bright228 or has a ‘dirty grey’ tinge.229 Less frequently, patients report a tinge to the world, as if peering through a colored filter.230 Daily activities that use color discrimination are impaired, such as distinguishing coins, stamps, or traffic lights: A good account of the experience of an achromatopsic artist has been published.231 Testing in these subjects showed deficits in the discrimination of hue (Figure 22-10) and saturation.
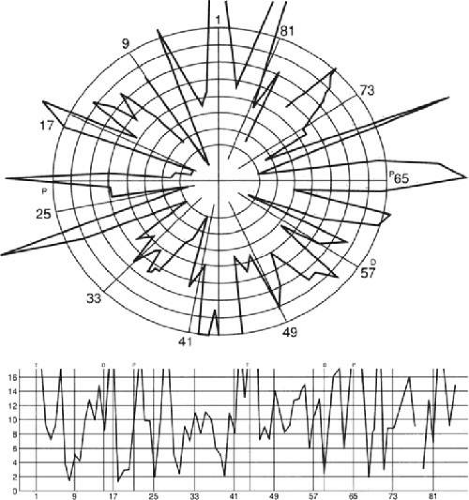 Figure 22-10. FM 100-Hue Test evaluates just noticeable differences around a hue circle in 85 equidistant steps at isoluminance. Different sectors of the rack are tested with four different racks consisting of 21 or 22 colors each. Plotting the errors produces a graph with a starburst appearance in a patient with severe cerebral chromatopsia. |
Effects on color constancy are an important issue. The wavelengths reaching the eye from an object depend on both its reflectant properties and the light illuminating the scene. Despite this, the color of objects remains stable under a wide range of environmental and lighting conditions.232,233 For instance, an apple continues to look red in sunlight, incandescent light, and fluorescent light, in an orchard or in a grocery display. Discounting the illuminant depends on computations in retina and cortex that average the spectral luminance over large regions of the surrounding background to infer the illuminant, which is then taken into account to derive the true color of an object.232,234 A defect in color constancy should result in color percepts that vary with illumination. This can be evaluated in dyschromatopsic patients, who have some residual hue sensitivity: Studies have shown that their color constancy is indeed impaired.235,236,237,238
Not all color perception is lost in achromatopsia. Some color processing of the cones and retinal ganglion cells of the parvocellular pathway can still be discerned in testing. Both trichromacy and color opponency have been shown in photopic spectral sensitivity curves235,239,240 and evoked potential or psychophysical measures of chromatic contrast sensitivity.240,241 Likewise, performance on anomaloscope tests can resemble an anomalous trichromat rather than a monochromat, despite the experience of the world as monochromatic shades of grey or sepia-tints.242,243 Achromatopsic patients can use color-opponent signals to detect a difference between colors and locate boundaries between differently colored regions of an image, even though they do not know what the colors are and cannot order them correctly by hue.244 This local chromatic contrast can help some patients detect the movement of chromatic stimuli245,246,247 and generate pupillary responses to color.248
Achromatopsia results from bilateral lesions of ventral occipitotemporal cortex. Lesions are often extensive, with other deficits such as superior quadrantanopia, prosopagnosia, and topographic disorientation. Superior field loss is almost always present, as the ventral occipitotemporal lesion that causes achromatopsia frequently extends into the inferior calcarine cortex or optic radiation. Similarly, only a few cases without prosopagnosia have been reported.249 Experimental testing has revealed a problem with detecting stimuli with low salience in some patients,250 which is thought to indicate inefficient attentional allocation in form processing. Other occasionally associated defects include visual object agnosia,239,251 alexia in achromatopsic patients with right hemianopia,227,249 and amnesia with additional ventral temporal lobe damage.227,251
In humans, functional imaging reveals several anatomically distinct areas involved in color processing, notably a V4 homologue and a second area named V4-alpha or V8 in the fusiform gyrus,252,253 as well as other more distant regions.254,255,256 Thus color processing involves a network of regions, and it is probable that a severe achromatopsic defect may require damage to or disconnection of several components of the network, rather than just a lesion of a single region like the human V4 homologue.257,258,259 The depth of the color loss in turn seems to depend on the extent of the lesion, with smaller lesions resulting in dyschromatopsia and larger lesions in achromatopsia.260
Achromatopsia is most often a result of strokes. Bilateral sequential or simultaneous infarctions in the territories of both posterior cerebral arteries can occur, or multiple infarcts may occur with a coagulopathy.261 Other bilateral lesions causing achromatopsia include herpes simplex encephalitis,239 cerebral metastases,249 repeated focal seizures,262 focal dementia,263 and even migraine aura, causing a transient achromatopsia.264 Temporooccipital white matter damage has caused a reversible dyschromatopsia in one patient with carbon monoxide poisoning.265
Hemiachromatopsia
Loss of color perception in the contralateral hemifield can follow unilateral right or left occipital lesions. Patients are typically asymptomatic until the defect is demonstrated to them.223,224 Hemiachromatopsia is usually associated with a superior quadrantanopia223,224,229; therefore, the color defect is demonstrable only in the remaining inferior quadrant. The preserved color vision in the ipsilateral hemifield allows normal or near-normal performance on centrally viewed tests of color vision such as pseudo-isochromatic plates. The incidence of hemiachromatopsia is probably underestimated, given its asymptomatic nature and the failure of routine clinical color tests to detect its presence.
Color Anomia and Agnosia
Although patients with achromatopsia cannot discriminate hue and saturation, some can name colors; in contrast, those patients with color anomia and color agnosia can discriminate colors accurately but cannot name them. Such patients may not be aware of their deficits.
Color anomia may occur as part of a more general anomia in aphasic patients or as an isolated entity. Several types of specific color anomia have been described, all with left occipital lesions and often associated with right homonymous hemianopia, rather than the upper quadrant defects of achromatopsia. When it accompanies pure alexia and right homonymous hemianopia, color anomia may be part of an interhemispheric visual-verbal disconnection.266,267,268,269 Disruption of callosal connections in the splenium between the intact right striate cortex and the left angular gyrus prevents accurately perceived colors in the remaining left hemifield (right occipital lobe) from gaining access to language processors in the left hemisphere. Thus these subjects cannot read words or name colors that they see accurately in the left hemifield. On the other hand, some but not all of these subjects can name visual objects. Preservation of object naming in these subjects has led to the hypothesis that, unlike colors, objects can activate not only visual but also somesthetic representations of object shape, and that these representations could be transferred to the left hemisphere through more anterior corpus callosum connections.267
In color dysphasia, patients not only are unable to name visible colors but also are unable to name the colors of familiar objects they see or imagine, tasks that patients with the disconnection type of color anomia can do well.268 This defect is attributed to loss of an internal lexicon of colors. Most have left angular gyrus lesions, with associated alexia and agraphia, right hemifield defects, and Gerstmann syndrome.
Color agnosia is also rare.270,271,272 These patients can sort and match colors, and some can even name colors they see. However, they cannot color line drawings of objects correctly or state whether objects have been colored correctly by others. They cannot learn paired associations between seen objects and seen colors (a visual-visual task), but, in contrast to patients with color amnesia, they can remember the stereotyped colors linked to certain objects, like the red of a fire engine or the yellow of a banana.273 Associated defects include pure alexia, object anomia, and poor performance on imagery tests for other object properties. Most of these subjects had left occipitotemporal lesions, some with a right hemianopia. A rare developmental form has also been described, with autosomal dominant inheritance.274
VISUAL AGNOSIA
Patients with visual agnosia no longer recognize previously familiar objects and cannot learn to identify new objects by sight alone.275,276 A historical debate centers on the necessary and sufficient impairments that generate agnosia and the extent to which these impairments involve memory rather than perception. Teuber277 defined agnosia as an associative disorder in which percepts are stripped of their meanings. This associative agnosia can be considered a selective disturbance of visual memory. In contrast, perceptual dysfunction is the main cause of disordered visual recognition in apperceptive agnosia.278 However, this apperceptive/associative distinction is rarely encountered in a pure form: Most patients with agnosia have a combination of impairments of visual perception and memory.
General visual agnosia is a rare disorder in which patients cannot distinguish between objects at a very elementary level. Thus they mistake their wives for hats. Apperceptive forms tend to occur with bilateral posterior occipital lesions, with some classic cases a result of carbon monoxide poisoning.279,280 Associative visual agnosia may reflect semantic access problems and sometimes show interesting dissociations between object categories, as between living and non-living objects or between objects and actions.
Selective visual agnosias are also uncommon but have received much interest because of the implications for anatomic localization and the modularity of visual cortex. These include prosopagnosia and alexia.
Prosopagnosia
Prosopagnosia is the impaired ability to recognize familiar faces or to learn facial identity.281 Although impaired face recognition can be part of more generalized problems of perception, cognition, and memory, as in macular degeneration,282 Alzheimer,283,284,285 Huntington,286 and Parkinson disease,287,288 the term prosopagnosia should be reserved for cases with selective deficits in face recognition: that is, where the problem recognizing faces is disproportionately severe compared to other visual or cognitive dysfunction.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


