The spectrum of vesiculobullous eruptions of the oral cavity is wide and rich, with different disease entities that encompass different etiologies, pathogenesis, clinical manifestations, treatment plans, and prognostic ends. Trying to present all these entities in a comprehensive fashion is challenging, but in this article, most of the important entities pertaining to this topic have been encompassed in a concise manner.
Causes of vesiculobullous eruptions in the oral cavity encompass many entities that are usually of autoimmune- or immune-mediated etiology. Correlation of the clinical and immunologic findings is essential for the accurate classification and diagnosis and management of these cases. In this ensuing article, these entities are discussed in a classification that mainly reflects the prevailing histologic appearance of the different disease processes.
Oral intramucosal bullous diseases (pemphigus)
Definition
Pemphigus is a group of rare chronic autoimmune disorder characterized by blistering of the stratified squamous epithelium of the skin and mucosal surfaces and encompasses a number of subtypes, which include pemphigus vulgaris (PV) and pemphigus vegetans (PVeg), both commonly involve the oral mucosa; immunoglobulin (Ig) A pemphigus that also manifests oral lesions; and pemphigus foliaceous and its variant pemphigus erythematosus (Senear-Usher syndrome), which are rarely manifested orally and are primarily cutaneous.
Paraneoplastic pemphigus (PP), usually seen in association with lymphoproliferative diseases, is another important variant that can affect the oral cavity as well as drug-induced pemphigus.
Typically, a person develops a single variant of pemphigus, but cases of transition to other variants have been described.
Pemphigus Vulgaris
Incidence
PV is rare with an incidence of only 0.1 to 0.5 cases per 100,000 persons per year worldwide; however, it is the most common pemphigus variety affecting the oral mucosa. This disorder manifests in the fifth to sixth decades of life, with a slight female predilection. Rare juvenile cases have also been reported. PV has strong genetic associations with certain ethnic groups (Ashkenazi Jews and people of Mediterranean and South Asian origin).
Clinical presentation
Oral involvement is the hallmark of PV and is seen in 90% of patients ( Fig. 1 ). Indeed, 50% to 70% of cases present primarily with oral disease long before cutaneous manifestations appear (>1 year). However, those presenting with cutaneous manifestations also eventually show oral manifestations. All other body mucosal sites (including pharyngeal, esophageal, nasal, vaginal, rectal, and urinary) can also be involved with the disease.
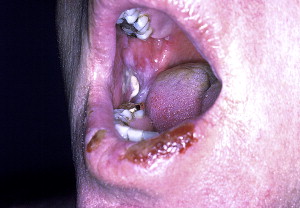
Lesions present as flaccid fluid-filled blisters or bullous lesions that eventually break, forming red, painful, erosive, irregular, ragged-edge, and ulcerated lesions. The buccal, palatal, ventral aspects of tongue and lip mucosa are commonly affected, and wide oral mucosa involvement can be seen in severe cases.
Gingival lesions are uncommon at the onset but may frequently appear on gingiva as isolated blisters and erosions mainly located in the free gingiva and are hardly recognizable as bullous lesions. However, in advanced cases, erosive or desquamative gingivitis is seen. If left untreated, new bullae forms as the older ones rupture; however, at the onset of the disease, these lesions present in a recurrent pattern and become persistent with time. Ulcers heal slowly, and scarring is rare.
These lesions translate themselves into typical symptoms of odynophagia, dysphagia, and severe discomfort experienced by the patient. A positive Nikolsky sign (slight rubbing of the skin or mucous membrane can lead to bullae formation or sloughing of the squamous epithelium) can be elicited in the mucous membrane or skin of affected individuals.
PV is a serious disease that is potentially fatal if blistering forms large denuded areas on the skin and mucous membranes. If left untreated, this condition has a mortality rate of 50% at 2 years, which reaches almost 100% at 5 years.
PV may be seen in association with other autoimmune disorders, including rheumatoid arthritis, myasthenia gravis, lupus erythematosus, and pernicious anemia.
Etiology
The causative factors in PV are largely idiopathic; however, some precipitating factors have been identified, including
- 1.
Genetic factors. As mentioned earlier, certain ethnic groups have a particular predilection for the disease. Associations with HLA antigen have also been recognized in these groups, particularly HLA-0DR4 (DRB1∗0402) in the Ashkenazi Jewish population, Europeans, and Asians with DRw14 (DRB1∗1041) and DQB1∗0503.
- 2.
Diet. Garlic in particular has been identified as an important dietary factor.
- 3.
Drugs. Drugs are a rare cause of PV, which include angiotensin-converting enzyme inhibitors (eg, captopril, enalapril, fosinopril), thiols (eg, cephalosporins, penicillamine, penicillin), antiinflammatory drugs (eg, aspirin, nonsteroidal antiinflammatory drugs [NSAIDs]), and a variable array of drugs (eg, levodopa, nifedipine, propranolol, rifampicin). Traditional cosmetic substances have been implicated in Tunisia as a cause of pemphigus.
- 4.
Viruses. The role of viruses in PV has been proposed mainly in relation to human herpesviruses (HHVs), particularly HHV-8 ; however, no confirmed role has been found yet.
- 5.
Several other factors have been implicated to cause PV, including smoking and exposure to pesticides. A role of increased estrogen hormone has also been suggested.
Pathogenesis
Loss of squamous cell-cell adhesion through damaged or defective desmosomal proteins, leading to vesiculation, bullae formation with subsequent rupture, and formations of erosions/ulcers, broadly describes the sequence of the pathogenic events in PV.
The oral squamous epithelium is similar to its skin counter part, however, with different desmosomal components. The oral epithelium expresses mainly cadherin-type cell adhesion molecule desmoglein 3 (Dsg3) that binds squamous cell to each other, whereas the skin expresses both Dsg1 and Dsg3 consequently, resulting in different disease manifestations. Patients with predominant oral manifestations express only Dsg3 antibodies. In oral PV, there is deposition of IgG class autoantibodies intercellularly against Dsg3. In patients with oral and cutaneous manifestations, the fact that the skin integrity is more maintained by Dsg1 than by Dsg3 makes the oral manifestations predominant until the Dsg1 antibodies also appear, resulting in severe cutaneous manifestations. In active disease, IgG4 autoantibodies are formed, and during remission, IgG1 predominates.
HLA class II alleles seem to be particularly important in the recognition of Dsg3 by T lymphocytes. The autoantibodies bind to Dsg3 and Dsg1 and form intercellular immune complexes that activate the complement by inflammatory mediators and activated T cells, resulting in damage to the intercellular junction, leading to cell apoptosis by proteinases, Fas, and caspase, eventually causing cleavage/blistering.
Histologic features
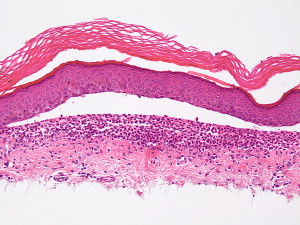
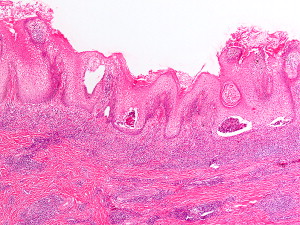
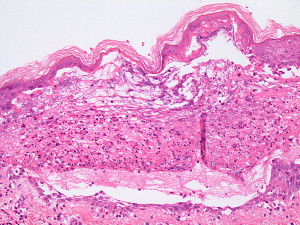
The hallmark of PV is the formation of bullae immediately above the basal cell layer (suprabasal bullae) ( Fig. 2 ). The sequence of events starts with slight intercellular edema within the epithelial layer with subsequent acantholysis low in the stratum spinosum, with cleft formations and eventually suprabasilar bullae formation.
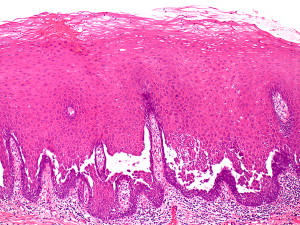
A single row of cuboidal basal cells remains below the bullae, giving the characteristic “tombstone” appearance ( Fig. 3 ). Papillae lined by a single layer of cuboidal cells and with a core formed by projecting submucosa are seen. No significant inflammation is seen early in the disease; however, focal eosinophilic infiltrate may be seen in the epidermis before acantholysis (eosinophilic spongiosis).
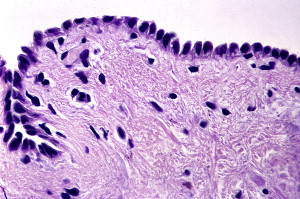
The formed acantholytic cells become hyperchromatic and swollen with high nuclear cytoplasmic ratio and are identified in smears collected from the lesions aiding in the lesion diagnosis. These cells are known as Tzanck cells.
Diagnosis
In addition to the clinical features, the following steps are used in the diagnosis of PV
- 1.
Perilesional tissue biopsy and histopathologic examination
- 2.
Direct immunofluorescence (DIF) microscopy or immunohistochemistry showing intercellular deposits of IgG and complement C3, which are more sensitive than conventional histopathology ( Fig. 4 )
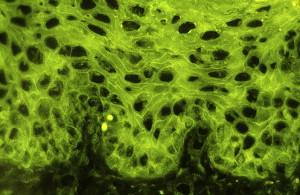
Fig. 4
A tissue biopsy result showing intercellular immunofluorescence with IgG in PV.
- 3.
Assays by indirect immunofluorescence (IIF) microscopy of serum antibody titers or by the use of specific enzyme-linked immunosorbent assays (ELISA).
Treatment and prognosis
Systemic immunosuppression is the mainstay of treatment to bring the disease process under control and to achieve complete and long-standing remission. With the proper immunosuppressive therapy, about half the patients achieve complete and long-standing remission after about 5 years.
Systemic corticosteroids seem to be the mainstay of treatments. Other treatments include plasmapheresis alone or in association with cyclosporin or cyclophosphamide (Kiel synchronization protocol). Others drugs include dapsone, chlorambucil, cyclophosphamide, azathioprine, gold, minocycline, and methotrexate. However, many of these drugs have adverse effects, and most clinicians focus on corticosteroid therapy.
Other emerging recent agents include intravenous immunoglobulins in steroid-resistant PV, mofetil, tacrolimus, and cholinergic agonists. Immunoadsorption and immunomodulation are other new treatment modalities.
Pemphigus Vegetans
Incidence
PVeg, a variant of PV, is a rare cause of pemphigus that constitutes 1% to 2% of all pemphigus cases. However, if this condition occurs, it almost always involves the oral cavity. The age of onset is usually between 40 and 50 years; however, any age group, including children, can be involved.
Clinical presentation
Similar to PV, more than half the cases of PVeg show oral manifestations preceding cutaneous ones and those with cutaneous lesions eventually have oral involvement.
PVeg has 2 types, Neumann and Hallopeau, of which the Neumann subtype is more common. Some studies have questioned this classification. In the Neumann type, the lesions are similar to those of PV. In the Hallopeau type, there are pustular lesions with a benign course and few relapses. Both lesions develop hyperpigmented verrucous vegetative plaques ( Fig. 5 ) with pustules and hypertrophic granulation tissue at the periphery. It is these verrucous vegetations that characterize PVeg and are painful, foul smelling in the cutaneous sites, and inflamed and vary in size from small lesions to larger-size areas. The propensity for bullae formation in PVeg is generally lower than that of PV.
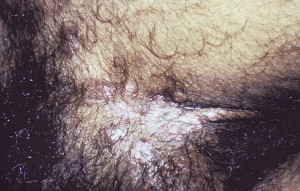
In addition to the oral mucosal lesions, the tongue seems to acquire a cerebriform appearance with numerous sulci and gyri. The cutaneous lesions are also typically seen in the intertriginous locations (eg, axilla, groin).
Pathogenesis and etiology
As in PV, autoantibodies are mainly formed against cell adhesion molecule Dsg 3. In cutaneous involvement, Dsg1 is also seen in a similar fashion to PV. Desmocollin 1 and 2 and periplakin have also been reported. The autoantibodies seen are IgG type, mostly IgG4 and, to a lesser degree, IgG1. A combination of IgA and IgG has also been reported in a rare case.
The etiopathogenesis of Pveg is not clear, captopril and intranasal heroine use has been reported.
Histologic features and diagnosis
The Neumann type shows histologic features similar to those seen in PV; however, eosinophilic spongiosis, in which the pustules seen in the early vegetations are filled with eosinophils, is a more prominent feature ( Fig. 6 ). Older vegetations ( Fig. 7 ) tend to demobstrate papillomatosis and hyperkeratosis and are less diagnostic.
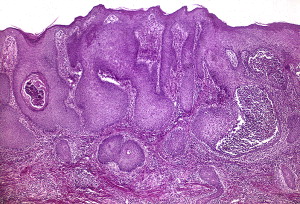
The Hallopeau type show shows similar changes; however, the intraepidermal eosinophilic abscesses formed tend to be larger and more numerous.
Diagnostic laboratory findings are similar to those of PV.
Treatment and prognosis
Systemic glucocorticoids are the treatment of choice. Oral administration of corticosteroids alone may not induce remission, and addition of immunosuppressive agents (eg, cyclophosphamide, azathioprine, etretinate) may improve remission rates.
Concerning prognosis, patients with Neumann type have a course similar to that of those with PV. Those patients with Hallopeau type have fewer, if any, relapses and usually respond to even lower doses of corticosteroids.
Paraneoplastic Pemphigus
Background
Anhalt and colleagues, in 1990, first described PP and put the original criteria for diagnosis, which includes
- •
Painful mucosa eruptions, with polymorphous skin eruptions in the setting of confirmed or occult malignancy
- •
Histopathologic changes ,such as acantholysis, keratinocyte necrosis, and interface dermatitis
- •
DIF observation of epidermal, intercellular, and basement membrane immunoreactants, mostly IgG and C3
- •
IIF observation of circulating antibodies directed against squamous, simple, columnar, and transitional epithelium
- •
Immunoprecipitation with a complex of 4 proteins (desmoplakin I, envoplakin, desmoplakin, and periplakin).
In 1993, Camisa and Helm also suggested a set of major and minor criteria. In 2004, Anhalt suggested minor criteria for diagnosis, including
- •
Painful progressive stomatitis
- •
Histopathologic changes of acantholysis or lichenoid dermatitis
- •
Demonstration of antiplakin antibodies
- •
Demonstration of an underlying neoplasm (mostly hematologic malignancies).
A recent literature review lists benign and malignant neoplasms that can be associated with PP, including Hematologic (84% of reported cases)
- •
Non-Hodgkin lymphoma (most frequent)
- •
Chronic lymphocytic leukemia
- •
Thymoma
- •
Castleman disease
- •
Waldenström macroglobulinemia
- •
Hodgkin lymphoma
- •
Monoclonal gammopathy
- •
Systemic mastocytosis.
Nonhematologic (16%)
- •
Carcinomas (squamous cell carcinoma of tongue, vagina, and skin; bronchogenic carcinoma; endometrial carcinoma)
- •
Sarcomas (liposarcoma, poorly differentiated sarcomas, leiomyosarcoma, dendritic cell sarcoma, malignant nerve sheath tumor).
Incidence
The exact incidence of PP is not clear from the existing literature, but an article in 2004 puts the number of reported cases at least at 150 cases. The mean age of occurrence is 60 years, with patients varying from ages 7 to 75 years. Men and women seem to be equally affected.
Clinical manifestations
Oral involvement is seen in 100% of reported cases. Extensive erosions and shallow ulcerations of the oral mucosa are usually seen. Involvement of the nasopharynx, tonsils, and esophagus in the head and neck region as well as conjunctival lesions may be observed. Initial presentation with oral lesions is present in 45% of cases. Cutaneous lesions have been reported in most cases and present in 5 different morphologies, such as pemphiguslike ( Fig. 8 ), pemphigoidlike, graft-versus-host disease (GVHD)-like, erythema multiforme–like, and lichen planus type. Other organ systems affected include the respiratory and the gastrointestinal tracts.
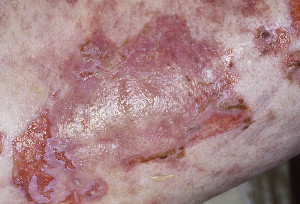
Pathogenesis and etiology
PP is thought to be caused by evoking both cellular and humoral immune response against tumor antigens that lead to blistering in the mucosal and epithelial surfaces. The antibodies formed are directed against desmoplakin I, desmoplakin II, periplakin, and envoplakin. Patients with PP also have antibodies to Dsg 3.
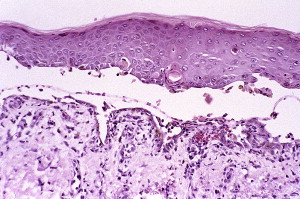
Histologic features and diagnosis
The histologic changes are similar to those observed in PV ( Fig. 9 ). However, changes similar to those seen in erythema multiforme have been reported. As mentioned earlier, the diagnosis depends on major and minor criteria; 3 major or 2 major and 2 minor criteria are required for the diagnosis. As in PV, immunoglobulins and complements are deposited in the epidermis in an intercellular pattern ( Fig. 10 ). The immunoprecipitation pattern is characteristic of PP.
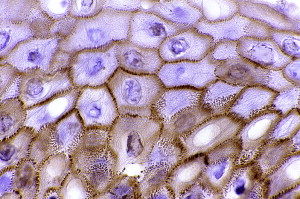
Treatment and prognosis
Treatment of PP is difficult, and the disease has poor prognosis with a survival rate of few months. Treatment of the original tumor may improve symptoms. Corticosteroid use, alone or in combination with other medications (eg, cyclosporines, cyclophosphamides, plasmapheresis, immunophoresis), has been reported with limited success.
IgA Pemphigus
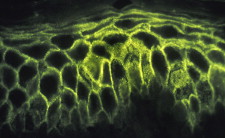
IgA pemphigus is a newly characterized, rare group of immune-mediated intraepidermal blistering skin ( Fig. 11 ) and mucous membrane diseases and is characterized by tissue-bound and circulating IgA autoantibodies. These diseases commonly affect the oral cavity. Histologically, epidermal acantholysis and neutrophilic infiltration predominates ( Fig. 12 ). The pathomechanisms of initiating the IgA autoantibodies to target desmosomal components ( Fig. 13 ) (Dsg3, desmocollin 1, and desmocollin 2) are not characterized yet. The clinical course of IgA-mediated pemphigus seems to be less aggressive than IgG-mediated pemphigus, with no significant morbidity.
Drug-Induced Pemphigus
Many of the drugs inducing pemphigus have been discussed under PV. Most of the patients generally improve and experience regression on cessation of the inducing drug.
Oral submucosal bullous diseases (pemphigoides)
Definition
Pemphigoid is a group of chronic immune-mediated subepithelial blistering diseases that includes cicatricial pemphigoid (CP), renamed mucous membrane pemphigoid (MMP), which is the most common form; bullous pemphigoid (BP); pemphigoid (herpes) gestationis; lichen planus pemphigoides; dermatitis herpetiformis (DH); linear IgA disease; epidermolysis bullosa acquisita (EBA); and bullous systemic lupus erythematosus (LE). Subepithelial bullous blistering has also been seen in PP-related cases.
In pemphigoid, IgG autoantibodies can be directed against various antigens of the epithelial basement membrane. In this article, the 2 most common types of pemphigoid that can affect the oral cavity, namely CP and BP, are addressed.
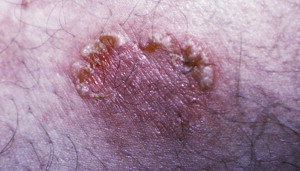
Cicatricial Pemphigoid (also known as Mucous Membrane Pemphigoid)
Incidence
CP is a rare condition that mainly affects the mucosal surfaces, particularly the oral mucosa ( Fig. 14 ). This condition usually affects women more than men (1.5:1 ratio), with an age range of 50 to 62 years according to some studies. However, the exact incidence and prevalence is unknown, but there does not seem to be a racial or geographic preference. Recent molecular characterizations of the disease have revealed the heterogeneity of CP.
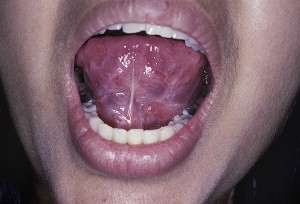
Clinical presentation
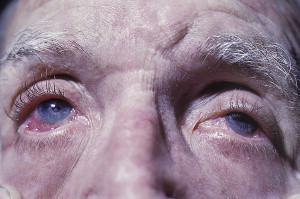
The heterogeneity of CP reflects itself in the variations of its clinical presentations. Patients can present with oral lesions, alone or with involvements of other mucous membranes or of skin, or systemic manifestations. Ocular involvement is a particularly serious complication ( Figs. 15 and 16 ).
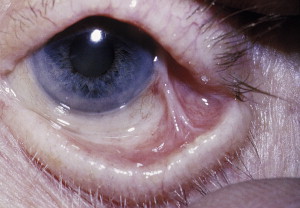
The oral mucosa is the initial site of lesions in most cases in which bullous or vesicular lesions can occur at any of its sites. Blisters and bullae eventually rupture, leaving irregularly shaped painful erosions, which sometimes results in scarring of the mucous membrane; hence the name cicatricial with the formation of adhesions in severe cases. If the gingiva is affected, desquamative gingivitis is seen, which varies from mild to severe forms.
Lesions can be limited to the oral cavity or can also affect the mucosa in other body sites (nose, esophagus, larynx, eyes, anus). Up to 40% of patients who present with oral CP can develop ocular disease that can result in severe conequences, such as scarring of the conjunctiva with scleral fusion, inverted eye lid (entropion), and trichiasis. These developments can all eventually lead to opacification and blindness. Skin involvement is usually seen in one-third of cases and can be limited to face, neck, scalp, axilla, trunk, and extremities. A Nikolsky sign, as observed with PV, can be seen with CP.
Association with other autoimmune disorders and also sometimes with B-cell lymphoproliferative disorders has been observed.
Pathogenesis and etiology
The initiating events for CP are not usually known. However, in some instances, a factor, such as, a drug may be the initiating agent (eg, a medication such as furosemide).
CP is associated with autoantibodies to several components of the basement membrane autoantigens, such as BP antigen (BPAg) 2 and less often to BPAg1, laminin 5 (epiligrin), laminin 6, type VII collagen, or β4 subunit of α 6 β 4 integrin. HLA-DQB1∗0301 may play a role in T-lymphocyte recognition of these antigens.
The autoantibodies formed are usually IgG and C3, but IgA or IgM may also be encountered, which may be a result of the heterogeneity of antigens.
Complement activation with resultant increase in cytokines and other enzymes along with basal cell detachment may be one of the mechanisms involved.
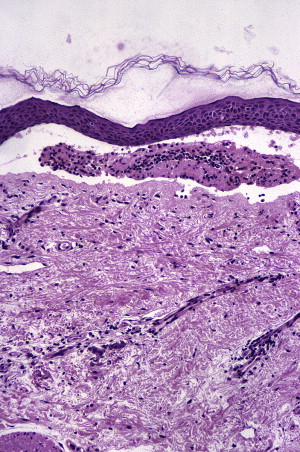
Histologic features and diagnosis
Subepithelial split with mild, chronic, inflammatory, cell infiltrate (lymphocytes, plasma cells, eosinophils, and neutrophils) may be observed ( Fig. 17 ). The tissue, as in all these cases, should be taken from the tissue next to the ulcerated area.
DIF studies show deposits, usually IgG and C3, in a homogeneous linear fashion in the basement membrane zone along the epitheliomesenchymal junction.
IIF and immunoblot assays are used to detect circulating antibodies. Using salt-split mucosa provides more sensitive assay results and shows IgG in the bullous floor. Other tests include direct and indirect immunoelectron microscopy, immunoblotting, immunoprecipitation, and ELISA.
Treatment and prognosis
Patients with only oral lesions have good prognosis and have been treated with topical drugs (corticosteroids, calcineurin anatagonists). Patients with lesions that are not limited to the oral cavity have a more problematic disease process and have been treated with systemic drugs, including dapsone, corticosteroids, azathioprine, cyclophosphamide, methotrexate, mycophenolate mofetil, sulfasalazine, and other drugs.
Patients who do not respond to the conventional treatments, such as high-dose systemic corticosteroids, immunosuppressant, or both, can be treated using intravenous immunoglobulins. Plasmapheresis has also been used in treating some patients. Surgical intervention is sometimes required to repair scars or other severe complications. However, surgical intervention should be advisably used because it may aggravate the disease.
Bullous Pemphigoid
Incidence
BP is uncommon in the United States, and its incidence is not clear. In Europe, this condition is recognized as the most common subepidermal bullous disease. It affects mostly patients older than 60 years, with equal sex predilection. Infant and childhood BP has also been reported. No definitive racial or ethnic link has been identified. Primarily a skin disease, BP can affect the mucosal surfaces in 10% to 40% of cases.
Clinical presentation
BP has been reported in the oral cavity in up to 40% of the patients in some series, but presence of the condition in the oral cavity is not usually the presenting symptom, and the presenting symptom is usually limited to the oropharynx. Most of the cases present with skin lesions; however, ocular involvement can also be seen rarely. The onset may be acute or subacute. Pruritus and widespread tense blisters are usually seen on an erythematous skin ( Fig. 18 ). The blisters are usually tense and eventually they rupture, become flaccid and ulcerate, resulting in an ulcerated erythematous area. The size of the blisters is variable from small vesicles to larger bullae.
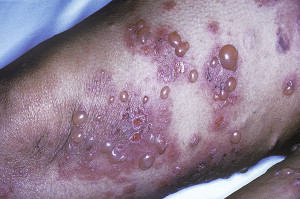
Several distinct clinical forms of BP occur, including
- •
Generalized bullous form, oral and ocular involvement usually occurs with this form
- •
Vesicular form
- •
Vegetative plaque form
- •
Generalized erythroderma form
- •
Urticarial form from which bullae eventually arise
- •
Nodular form
- •
Acral form
- •
Infant form.
Pathogenesis and etiology
BP has been reported to be induced by ultraviolet radiation, radiotherapy, and some drugs or medications furosemide, ibuprofen, and other NSAIDs, antibiotics, penicillamine, and captopril, among others.
In BP autoantibodies, usually IgGs are formed against specific hemidesmosomal BP antigens BP230 (BPAg1) and a transmembrane protein with a collagenous extracellular domain BP 180 (BPAg2). BPAg2 has been identified as the major antigen involved in the development of BP, and serum autoantibodies against BPAg2 has been correlated with the disease activity. Other autoantibodies against α 6 integrin and laminin 5 have also been observed.
Some cytokines and chemokines have been found to play a role in BP, including eotaxin, interleukin 16, MIG, CCL17, and CCL22.
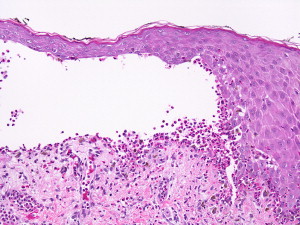
Histologic features and diagnosis
The samples for biopsies, like in other bullous diseases, are obtained from the perilesional tissue of a new blister. A classic biopsy specimen shows a subepidermal or submucosal bulla with a viable roof. The associated inflammatory infiltrate is mixed and is particularly rich in eosinophils, which is contained in the bullous cavity ( Fig. 19 ). Mast cells and basophils can predominate early in the course of the disease. A cell-poor BP with minimal cellular infiltrate has also been observed.
DIF studies usually demonstrate IgG and C3 deposition in a linear fashion ( Fig. 20 ) along the junction of the epithelium and subepithelium. DIF assay on a salt-split skin demonstrates IgG in the blister roof in patients with BP, which differentiates BP from CP and EBA.
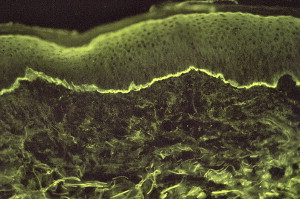
IIF and immunoblot assays are used to detect circulating antibodies. Other tests include direct and indirect immunoelectron microscopy, immunoblotting, immunoprecipitation, and ELISA.
Treatment and prognosis
In most patients with BP who are treated for the condition, the disease remits within 1.5 to 5 years. However, BP can be life threatening in debilitated patients. Treatment modalities include antiinflammatory agents (eg, corticosteroids, dapsone, tetracyclines) and immunosuppressant agents (eg, azathioprine, methotrexate, mycophenolate mofetil, cyclophosphamide). Topical corticosteroids have been advocated in place of systemic steroids to avoid the side effects of systemic treatment, particularly in the elderly.
Dermatitis Herpetiformis
Incidence
DH is an autoimmune disorder that is associated with gluten-sensitive enteropathy. DH occurs more frequently in the people of Northern European descent and is rare in Asians and Africans. The prevalence of DH is approximately 10 per 100,000 cases worldwide. HLA-DQA1∗0501, HLA-DR3, HLA-B8, and HLA-B1∗02, which encodes HLA-DQ2, heterodimers have been linked to prevalence. A male to female ratio of 1.5:1 or 2:1 is seen, usually affecting people mostly in the second to fourth decades of life. However, any age, including children’s age groups, may be affected.
Oral involvement in DH is extremely rare, and patients with linear IgA bullous dermatosis (discussed later) are more likely to have oral involvement.
Clinical presentation
The history is typically of intensely pruritic papulovesicular eruptions on the extensor surfaces of the arms, knees, back, and buttocks. Gluten, as mentioned, exacerbates symptoms. Periods of exacerbation and remissions are typical of the disease. Oral involvement, as mentioned, is infrequent and men are more likely affected than women. Observation of vesicles and bullae in the oral mucosa is rather difficult because these structures tend to rupture or excoriate quickly, leaving tender ulcers.
Pathogenesis and etiology
Etiologically, DH is considered as one of the manifestations of celiac disease. The presence of granular IgA deposits in normal-appearing perilesional skin is detected by DIF assay in 92.4% of patients. Most of these deposits are located in the dermal papillae. Of the cases, 10% show continuous granular deposits at the basement membrane in both papillary and nonpapillary lesions. C3 deposits can also be seen.
Histologic features and diagnosis
Accumulation of neutrophils in the dermal papillae are noted, which can eventually form papillary microabscesses that causes vacuolization, with separation and blister formation between the tips of the epidermis and the papillae. Coalesced blisters may give rise to larger tense blisters with an increased number of eosinophils in the blister. Biopsy samples should be taken from the perilesional mucosa rather than from the blister site for more accurate diagnosis.
Most patients with oral DH have a histologic evidence of enteropathy. In addition to the granular and linear IgA deposits observed in this disease, serum markers, such as IgA endomysial antibodies, can be seen.
Treatment and prognosis
DH is a lifelong disease with variable severity. Patients on a gluten-free diet and who can tolerate dapsone treatment usually have a good prognosis. Patients who also have gluten-sensitive enteropathies accompanying DH are at risk of developing lymphoma.
Linear IgA Disease
Incidence
Linear IgA disease is a rare autoimmune subepithelial disease that predominantly involves the skin with bimodal presentation (age group of 6 months to 10 years in children and 14–83 years in adults); however, oral involvement has been reported in this entity in up to 70% of patients with the disease. The incidence of the disease has been only sporadically reported in different parts of the world; for example, the incidence in Utah (United States) is 0.6 per 100,000 cases and in France is 0.13 per 100,000 cases. A slight female predominance has been reported (1.6:1).
According to a recent review article, approximately 17 cases predominantly with oral involvement are reported in the literature, with the age range of these cases being between 29 and 79 years (10 women and 7 men). This finding suggests that IgA disease can manifest primarily as an oral presentation and not only second to a predominant skin presentation.
Clinical presentation
In most cases of linear IgA disease, the dermatologic manifestations are the main ones, with clear and/or hemorrhagic round or oval vesicles or bullae on normal, erythematous, or urticarial skin; erythematous papules; or blanching macules and papules. When this is the case, the oral manifestations tend to be minor. However, when the oral manifestations are the predominant, they include vesicles and bullae, painful ulcers and erosions, erosive cheilitis, desquamative gingivitis, and white patches with erythema, and these manifestations may be severe enough to be histologically and clinically confused with other entities, especially CP and erosive lichen planus. In the 17 reported cases of predominant oral presentation, gingival erythema and desquamative gingivitis were observed. Ocular lesions also occur.
Pathogenesis and etiology
The cause of the disease is related to an environmental trigger, such as infectious agents (eg, herpesvirus, varicella, and agents causing typhoid and upper respiratory tract infections), or drug (eg, vancomycin, captopril, ampicillin sodium, phenytoin, amoxicillin, cyclosporines) or can be associated with lymphoproliferative disease (eg, Hodgkin and non-Hodgkin lymphomas, chronic lymphocytic leukemia) and different types of cancers (eg, thyroid, colon, uterine) that lead to the production of IgA autoantibodies against different antigenic sites in the basement membrane, including the lamina lucida, sublamina densa, or both locations, and formation of the characteristic linear IgA deposits. These antigenic sites include a 97-kDa protein antigen, which may be the extracellular portion of BPAg2. Other antigens include a 120-kDa antigen, a 230-kDa antigen (BPAg1), and a 250-kDa antigen.
Histologic features and diagnosis
The early changes show neutrophilic infiltrate aligned along the basement membrane ( Fig. 21 ), with basal vacuolar degeneration. Neutrophilic microabscesses may be encountered in the dermal papillae. As the lesion progresses, subepidermal separation and blister formation without necrosis of the epithelial roof of the blister is seen, with predominantly polymorphic infiltrate that may show eosinophils.