Uveitis
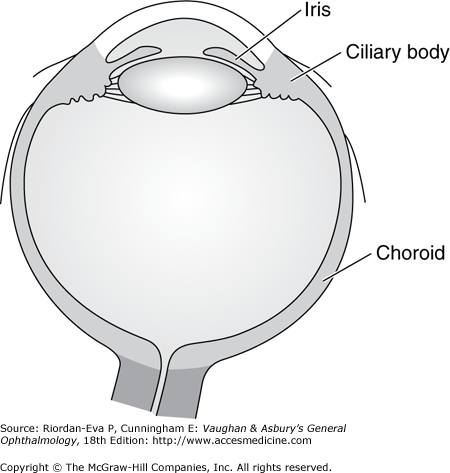
The uveal tract consists of the choroid, ciliary body, and iris (Figure 7–1). The term “uveitis” denotes inflammation of the iris (iritis, iridocyclitis), ciliary body (intermediate uveitis, cyclitis, peripheral uveitis, or pars planitis), or choroid (choroiditis). Common usage, however, includes inflammation of the retina (retinitis), retinal vasculature (retinal vasculitis), and intraocular optic nerve (papillitis). Uveitis may also occur secondary to inflammation of the cornea (keratitis), sclera (scleritis), or both (sclerokeratitis). Uveitis usually affects people 20–50 years of age and accounts for 10%–20% of cases of legal blindness in developed countries. Uveitis is more common in the developing world than in the developed countries, due in large part to the greater prevalence of infections that can affect the eye, such as toxoplasmosis and tuberculosis.
Inflammation of the uveal tract has many causes and may involve one or more regions of the eye simultaneously. Anatomically, intraocular inflammation can be classified as anterior uveitis, intermediate uveitis, posterior uveitis, or diffuse or panuveitis (Figure 7–2).
| Nongranulomatous | Granulomatous | |
|---|---|---|
| Onset | Acute | Insidious |
| Pain | Marked | None or minimal |
| Photophobia | Marked | Slight |
| Blurred vision | Moderate | Marked |
| Circumcorneal flush | Marked | Slight |
| Keratic precipitates | Small white | Large gray (“mutton fat”) |
| Pupil | Small and irregular | Small and irregular (variable) |
| Posterior synechiae | Sometimes | Sometimes |
| Iris nodules | None | Sometimes |
| Site | Anterior | Anterior, posterior, or diffuse |
| Course | Acute | Chronic |
| Recurrence | Common | Sometimes |
Figure 7-2.

Anatomic classification of uveitis, including anterior uveitis, intermediate uveitis, posterior uveitis and diffuse, or panuveitis. [Modified after Cunningham ET Jr. Diagnosis and management of acute anterior uveitis. American Academy of Ophthalmology, Focal Points 2002, Volume XX, Number 1 (Section 1 of 3).]
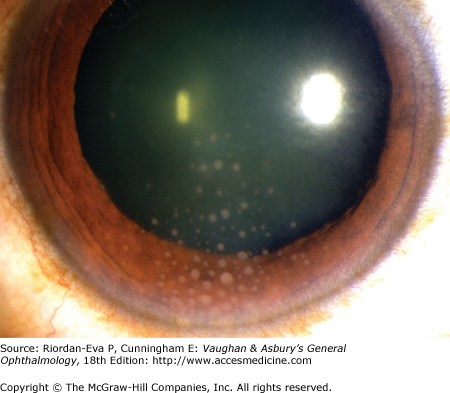
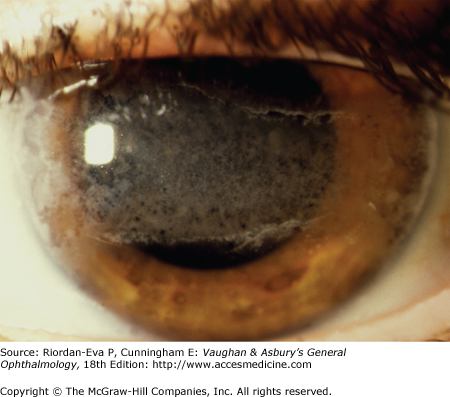
Anterior uveitis is most common and is usually unilateral and acute in onset. Typical symptoms include pain, photophobia, and blurred vision. Examination usually reveals circumcorneal redness with minimal injection of the palpebral conjunctiva or discharge. The pupil may be small (miosis) or irregular due to the formation of posterior synechiae. Inflammation limited to the anterior chamber is called “iritis,” whereas inflammation involving both the anterior chamber and the anterior vitreous is often referred to as “iridocyclitis.” Corneal sensation and intraocular pressure should be checked in every patient with uveitis. Decreased sensation occurs in patients with herpetic uveitis due to simplex or varicella-zoster virus infection or leprosy (see Chapter 15), whereas increased intraocular pressure can occur with herpes simplex virus, varicella-zoster virus, cytomegalovirus, toxoplasmosis, syphilis, sarcoidosis, or an uncommon form of iridocyclitis called glaucomatocyclitic crisis, also known as the Posner–Schlossman syndrome. Clumps of white cells and inflammatory debris termed keratic precipitates are usually evident on the corneal endothelium in patients with active inflammation. Keratic precipitates may be large so-called “mutton fat” or “granulomatous” (Figure 7–3), small and nongranulomatous, or stellate. Granulomatous or nongranulomatous keratic precipitates are usually located inferiorly in a wedge-shaped region known as Arlt’s triangle. Stellate keratic precipitates, in contrast, are usually distributed evenly over the entire corneal endothelium and may be seen in uveitis due to herpes simplex virus, varicella-zoster virus, cytomegalovirus, toxoplasmosis, Fuchs’ heterochromic iridocyclitis, and sarcoidosis. Keratic precipitates may also be localized to an area of prior or active kerititis, most frequently in herpetic keratouveitis. Iris nodules may be present at the iris margin (Koeppe nodules), within the iris stroma (Busacca nodules), or in the anterior chamber angle (Berlin’s nodules). Evidence for granulomatous disease, such as mutton fat keratic precipitates or iris nodules, may indicate an infectious cause of uveitis or one of a relatively limited number of noninfectious causes, including sarcoidosis, Vogt–Koyanagi–Harada disease, sympathetic ophthalmia, or lens-induced uveitis. Particularly severe anterior chamber inflammation may result in layering of inflammatory cells in the inferior angle (hypopyon). The most common cause of hypopyon uveitis in North America and Europe is HLA-B27–associated uveitis, whereas the most common cause of hypopyon uveitis in Asia is Behçet’s disease. The iris should be examined carefully for evidence of atrophy or transillumination, which can occur in a sectoral or patchy pattern in the setting of herpetic uveitis, or diffusely with Fuchs’ heterochromic iridocyclitis, also known as Fuchs uveitis syndrome. The presence of anterior (Figure 7–4) or posterior (Figures 7–5 and 7–6) synechiae should also be noted, as this can predispose the patient to ocular hypertension or glaucoma.
Intermediate uveitis, also called cyclitis, peripheral uveitis, or pars planitis, is the second most common type of intraocular inflammation. The hallmark of intermediate uveitis is vitreous inflammation. Intermediate uveitis is typically bilateral and tends to affect patients in their late teens or early adult years. Men and women are affected equally. Typical symptoms include floaters and blurred vision. Pain, photophobia, and redness are usually absent or minimal, although these symptoms may be more prominent at onset. The most striking finding on examination is vitritis, often accompanied by vitreous condensates, either free-floating as “snowballs” or layered over the pars plana and ciliary body as “snow-banking.” Mild anterior chamber inflammation may be present, but if significant, the inflammation is more appropriately termed diffuse uveitis or panuveitis (see later in the chapter). The cause of intermediate uveitis is unknown in the vast majority of patients, although sarcoidosis and multiple sclerosis account for 10%–20% of cases. Syphilis and tuberculosis, although uncommon, should be excluded in all patients. The most common complications of intermediate uveitis include cystoid macular edema, retinal vasculitis, and neovascularization of the optic disk and retina.
Posterior uveitis includes retinitis, choroiditis, retinal vasculitis, and papillitis, which may occur alone or in combination. Symptoms typically include floaters, loss of visual field or scotomas, or decreased vision, which can be severe. Retinal detachment, although infrequent, occurs most commonly in posterior uveitis and may be tractional, rhegmatogenous, or exudative in nature.
Laboratory testing is usually not required for patients with mild uveitis and a recent history of trauma or surgery—or with clear evidence of herpes simplex or herpes zoster virus infection, such as a concurrent vesicular dermatitis, dendritic or disciform keratitis, or sectoral iris atrophy. Laboratory testing can also be deferred for otherwise healthy and asymptomatic young to middle-aged patients with a first episode of mild to moderately severe acute, unilateral, nongranulomatous iritis or iridocyclitis that responds promptly to treatment with topical corticosteroids and cycloplegic/mydriatic agents. Patients with recurrent, severe, bilateral, granulomatous, intermediate, posterior, or panuveitis should be tested, however, as should any patient whose uveitis fails to respond promptly to standard therapy. Testing for syphilis should include both a Venereal Disease Research Laboratory (VDRL) or rapid plasma reagin (RPR) test and a more specific test for anti-Treponema pallidum antibodies, such as the FTA-ABS or MHA-TP assays. Sarcoidosis should be excluded by chest x-ray and serum angiotensin-converting enzyme (ACE) or lysozyme level testing. Tuberculosis should be excluded by the same chest x-ray, and by either skin testing using both purified protein derivative (PPD) or by an interferon-γ release assay (IGRA), such as the QuantiFERON-TB Gold or T-SPOT.TB tests. While the IRGAs provide markedly increased specificity for patients with prior BCG vaccination, a remote history of BCG vaccination should not preclude PPD skin testing in areas where IRGAs are not available, since the PPD test should become negative (<5 mm induration) within 5 years following BCG vaccination. Testing other than for syphilis, tuberculosis, and sarcoidosis should be tailored to findings elicited on history or identified on physical examination. Examples might include an antinuclear antibody (ANA) titer for a young child with chronic iridocyclitis and arthritis suspected of having juvenile idiopathic arthritis (JIA); an HLA-B27 histocompatibility antigen test for patients with arthritis, psoriasis, urethritis, or symptoms consistent with inflammatory bowel disease; or toxoplasmosis IgG and IgM titers for a patient with unilateral panuveitis and focal retinochoroiditis.
The differential diagnosis for eye redness and decreased vision is extensive and somewhat beyond the scope of this brief overview. However, entities commonly confused with uveitis include conjunctivitis, distinguished by the presence of discharge and redness involving both the palpebral and bulbar conjunctiva; keratitis, distinguished by the presence of epithelial staining or defects or by stromal thickening or infiltrate; and acute angle closure glaucoma, associated with markedly elevated intraocular pressure, corneal haziness and edema, and a narrow anterior chamber angle, often best visualized in the uninvolved fellow eye. (see Differential Diagnosis of Common Causes of the Inflamed Eye.)
Anterior uveitis can produce both anterior (Figure 7–4) and posterior synechiae (Figures 7–5 and 7–6)). Anterior synechiae can impede aqueous outflow at the chamber angle and cause ocular hypertension or glaucoma. Posterior synechiae, when extensive, can cause secondary angle closure glaucoma by producing pupillary seclusion and forward bulging of the iris (iris bombé). Early and aggressive use of corticosteroids and cycloplegic/mydriatic agents lessens the likelihood of these complications.

Both anterior and posterior chamber inflammation promote lens thickening and opacification. Early in the course, this can cause a simple shift in refractive error, usually toward myopia. With time, however, cataract progression often limits best-corrected vision. Treatment involves removal of the cataract, but should be done only when the intraocular inflammation is well controlled for at least 6 months, since the risk of intraoperative and postoperative complications is greater in patients with active uveitis. Aggressive use of local and systemic corticosteroids is usually necessary before, during, and after cataract surgery in these patients.
Cystoid macular edema is a common cause of visual loss in patients with uveitis and may be observed in the setting of severe anterior or intermediate uveitis. Longstanding or recurrent macular edema can cause permanent loss of vision due to cystoid degeneration. Both fluorescein angiography and optical coherence tomography can be used to diagnose cystoid macular edema and to monitor its response to therapy.
Retinal detachments, including tractional, rhegmatogenous, and exudative forms, occur infrequently in patients with posterior, intermediate, or panuveitis. Exudative retinal detachment suggests significant choroidal inflammation and occurs most commonly in association with Vogt-Koyanagi-Harada disease, sympathetic ophthalmia, and posterior scleritis or in association with severe retinitis or retinal vasculitis.
Corticosteroids and cycloplegic/mydriatic agents are the mainstays of therapy for uveitis. Care should be taken to rule out an epithelial defect and ruptured globe when a history of trauma is elicited and to check corneal sensation and intraocular pressure to rule out herpes virus infection. Aggressive topical therapy with a potent corticosteroid, such as 1% prednisolone acetate, one or two drops in the affected eye every 1 or 2 hours while awake, usually provides good control of anterior inflammation. Prednisolone acetate is a suspension and needs to be shaken vigorously prior to each use. A cycloplegic/mydriatic agent, such as homatropine 2 or 5%, used two to four times daily, helps prevent synechia formation and reduces discomfort from ciliary spasm.
Noninfectious intermediate, posterior, and panuveitis responds best to sub-Tenon injections of triamcinolone acetonide, usually 1 mL (40 mg) given superotemporally. Intraocular triamcinolone acetonide, 0.05–0.1 mL (2–4 mg), or oral prednisone, 0.5–1.5 mg/kg/d, can also be effective. Corticosteroid-sparing agents such as methotrexate, azathioprine, mycophenolate mofetil, cyclosporine, tacrolimus, cyclophosphamide, chlorambucil, or the TNF-α inhibitors can be required to treat severe or chronic forms of noninfectious inflammation, particularly when there is systemic involvement. Therapy for selected granulomatous forms of uveitis is outlined in Table 7–2.
| Anti-infective Chemotherapy | Use of Corticosteroids | |
|---|---|---|
| Toxoplasmosis | If central vision is threatened, give pyrimethamine, 75 mg orally as a loading dose for 2 days followed by 25–50 mg once daily for 4 weeks, in combination with trisulfapyrimidines (sulfadiazine, sulfamerazine, and sulfamethazine, 0.167 g of each per tablet), 2 g orally as loading dose followed by 0.5–1 g 4 times daily for 4 weeks. If a fall in the white blood cell or platelet count occurs during therapy, give folinic acid (leucovorin), 1 mL IM twice weekly or 3 mg orally twice weekly. Alternative chemotheraputic approach for ocular toxoplasmosis: Clindamycin, 300 mg orally 4 times a day with sulfonamides (as above), or spiramycin 1g 3 times daily, or minocycline, 100 mg orally daily for 3–4 weeks. | If the response is not favorable after 2 weeks, continue the anti-infective therapy and give systemic corticosteroids, eg, prednisone, 0.5 mg/kg/d with tapering over 3–4 weeks. Never stop anti-infective therapy prior to stopping corticosteroids. |
| Tuberculosis | Isoniazid, 300 mg orally daily, rifampicin, 450–600 mg orally daily, and pyridoxine, 50 mg orally daily, for 6–9 months; with ethambutol, 15 mg/kg orally daily and pyrazinamide 1.5–2 g orally daily for initial 2 months. | If a favorable response does not occur in 6 weeks, continue antimycobacterial therapy and give systemic corticosteroids, eg, prednisone, 0.5–1 mg/kg/d, with tapering as allowed by response. |
| Sarcoidosis | Treat with local corticosteroids, cycloplegic/mydriatic agents, and, as needed, with systemic corticosteroids such as prednisone, 0.5–1 mg/kg/d, with tapering as allowed by response. The usual contraindications to systemic corticosteroid therapy apply. A corticosteroid sparing agent is occasionally required | |
| Sympathetic ophthalmia | Treat with local corticosteroids, cycloplegic/mydriatic agents, and with systemic corticosteroids in high doses, eg, prednisone, 1–1.5 mg/kg/d. The usual contradictions to systemic corticosteroid therapy apply, and a corticosteroid sparing agent is often required. | |
Cataract and glaucoma are the most common complications of corticosteroid therapy. Cycloplegic/mydriatic agents weaken accommodation and can be particularly bothersome to patients under 45 years of age. Because oral corticosteroids or noncorticosteroid immunosuppressive agents can cause numerous systemic complications, dosing and monitoring are best done in close collaboration with an internist, rheumatologist, or oncologist.
The course and prognosis of uveitis depends to a large extent on the severity, location, and cause of the inflammation. In general, severe inflammation takes longer to treat and is more likely to cause intraocular damage and loss of vision than mild or moderate inflammation. Moreover, anterior uveitis tends to respond more promptly than intermediate, posterior, or panuveitis. Retinal, choroidal, or optic nerve involvement tends to be associated with a poorer prognosis.
About 20% of children with the pauciarticular form of JIA (formerly known as juvenile rheumatoid arthritis [JRA] in the United States and juvenile chronic arthritis [JCA] in the United Kingdom) develop a chronic bilateral nongranulomatous iridocyclitis. Girls are affected four to five times more commonly than boys. JIA-associated uveitis is usually detected at 5–6 years of age following the insidious onset of cataract (leukocoria), a difference in color of the two eyes (heterochromia), a difference in the size or shape of the pupil (anisocoria), or ocular misalignment (strabismus). Often these findings are first noted at a screening vision test performed at school. There is no correlation between the onset of the arthritis and that of the uveitis, which may precede the onset of arthritis by up to 10 years. The knee is the most commonly involved joint. The cardinal signs of the disease are cells and flare in the anterior chamber, small- to medium-sized white keratic precipitates with or without flecks of fibrin on the endothelium, posterior synechiae formation, often progressing to seclusion of the pupil, and cataract. Band keratopathy (Figure 7–7), secondary ocular hypertension or glaucoma, and cystoid macular edema can also be present and cause loss of vision. Patients suspected of having JIA should be evaluated by a rheumatologist and tested for a positive ANA titer.
| Autoimmune |
| Juvenile idiopathic arthritis |
| Ankylosing spondylitis |
| Reiter’s syndrome (reactive arthritis) |
| Inflammatory bowel disease (ulcerative colitis, Crohn’s disease) |
| Lens-induced uveitis |
| Sarcoidosis |
| Psoriatic arthritis |
| Infections |
| Syphilis |
| Tuberculosis |
| Leprosy (Hansen’s disease) |
| Herpes simplex virus |
| Varicella-zoster virus |
| Cytomegalovirus |
| Onchocerciasis |
| Leptospirosis |
| Malignancy |
| Masquerade syndrome |
| Retinoblastoma |
| Leukemia |
| Lymphoma |
| Malignant melanoma |
| Other |
| Idiopathic |
| Traumatic uveitis, including penetrating injuries |
| Retinal detachment |
| Fuchs’ heterochromic iridocyclitis (Fuchs’ uveitis syndrome) |
| Glaucomatocyclitic crisis (Posner-Schlossman syndrome) |
Treatment of JIA-associated uveitis is challenging. Topical corticosteroids, nonsteroidal anti-inflammatory agents, and cycloplegic/mydriatic agents are all of value. In resistant cases, systemic immunosuppression with non-corticosteroid immunosuppressive agents such as methotrexate, mycophenolate mofetil, or TNF-α inhibitors may be required to control the disease. Cataract surgery is associated with a relatively high risk of postoperative exacerbations, and intraocular lens implantation is usually contraindicated.
Up to 50% of patients with ankylosing spondylitis develop anterior uveitis. There is a marked preponderance for men. The uveitis can vary in severity from mild to severe and often produces pain, photophobia, and blurred vision. Limbal injection is usually present. Keratic precipitates, though usually present, are rarely granulomatous, and iris nodules do not occur. Posterior synechiae, peripheral anterior synechiae, cataracts, and glaucoma are common complications following severe, recurrent, or poorly controlled bouts of inflammation. Macular edema is uncommon, but can occur when the inflammation is severe and spills over to involve the vitreous. Recurrence is the rule and may involve either eye, although bilateral simultaneous involvement is atypical. The HLA-B27 histocompatibility antigen is present in approximately 50% of patients with acute nongranulomatous iritis or iridocyclitis seen in tertiary referral centers, but may be as high as 90% in community practice. Of those patients with anterior uveitis who are HLA-B27 positive, roughly half will experience a nonocular complication of their disease—most commonly ankylosing spondylitis, but Reiter’s syndrome (reactive arthritis), inflammatory bowel disease, and psoriatic arthritis may also occur. Imaging and colonoscopy can occasionally confirm diagnoses suspected on clinical grounds.
Fuchs’ heterochromic iridocyclitis is uncommon, accounting for less than 5% of all cases of uveitis. The onset is typically insidious during the third or fourth decade of life. Redness, pain, and photophobia tend to be minimal. Patients usually complain of blurred vision due to cataract. Iris heterochromia, best appreciated with natural lighting, can be subtle and is often most obvious over the iris sphincter muscle. Keratic precipitates are often small and stellate and scattered over the entire endothelium. Abnormal blood vessels may be seen in the chamber angle on gonioscopy. Posterior synechiae are uncommon, although they may occur in some patients following cataract surgery. A vitreous reaction may be present in 10% of patients. While loss of stromal pigment tends to make heavily pigmented eyes look hypochromic, stromal atrophy affecting lightly colored irides can actually reveal underlying pigment epithelium on the posterior surface of the iris, causing paradoxic hyperchromia. Pathologically, the iris and ciliary body show moderate atrophy with patchy depigmentation and diffuse infiltration of lymphocytes and plasma cells.
Cataract eventually develops in most patients, whereas glaucoma occurs in 10%–15% of cases. The prognosis is generally good. Cataract surgery can usually be performed without complication, and most patients with glaucoma can be managed with topical medications alone.
Lens-induced (phacogenic) uveitis is an autoimmune disease directed against lens antigens. There are no data at present to substantiate the suggestion that lens material per se is toxic, so the term “phacotoxic uveitis” should be avoided. The classic case occurs when the lens develops a hypermature cataract and the lens capsule leaks lens material into the posterior and anterior chambers. This material elicits an inflammatory reaction characterized by accumulation of plasma cells, mononuclear phagocytes, and a few polymorphonu-clear cells. Typical anterior uveitis symptoms of pain, photophobia, and blurred vision are common. Lens-induced uveitis may also occur following lens trauma or cataract surgery with retained lens material. Phacolytic glaucoma is a common complication. Definitive treatment requires removal of the lens material. Concurrent treatment with corticosteroids, cycloplegic/mydriatic agents, and intraocular pressure–lowering medications is often necessary.
Intermediate uveitis affects mainly the intermediate zone of the eye—ciliary body, principally the pars plana, peripheral retina, and vitreous. The cause is unknown in most cases, although syphilis, tuberculosis, and sarcoidosis should be ruled out with appropriate laboratory and ancillary testing. Multiple sclerosis should also be considered, particularly when supportive signs or symptoms are present. Intermediate uveitis is seen mainly among young adults, affects men and women equally, and is bilateral in up to 80% of cases. Common complaints include floaters and blurred vision. Pain, redness, and photophobia are unusual but can accompany a severe first attack. Adequate examination of the ciliary body, pars plana, and peripheral retina requires use of an indirect ophthalmoscope and scleral depression, which often reveals vitreous condensations in the form of snowballs and snowbanking. Adjacent retinal vasculitis is common. Anterior chamber inflammation is invariably mild, and posterior synechiae are uncommon. Posterior subcapsular cataract and cystoid macular edema are the most common causes of decreased vision. In severe cases, cyclitic membranes and retinal detachments may occur. Secondary glaucoma is rare. Corticosteroids are used mainly to treat cystoid macular edema or retinal neovascularization. Topical corticosteroids should be tried for 3–4 weeks to identify patients predisposed to development of corticosteroid-induced ocular hypertension. If no improvement is noted and ocular hypertension does not develop, a posterior sub-Tenon or intraocular injection of triamcinolone acetonide, 40 mg/mL, may be effective. Patients with intermediate uveitis usually do well with cataract surgery.
The retina, choroid, and optic nerve are affected by a variety of infectious and noninfectious disorders, the more common of which are listed in Table 7–4.
| Infectious disorders |
| Viruses |
| Cytomegalovirus, herpes simplex virus, varicella-zoster virus, rubella virus, rubeola virus |
| Bacteria |
| Agents of tuberculosis, brucellosis, sporadic and endemic syphilis; Borrelia (Lyme disease); and various hematogenously spread gram-positive and gram-negative pathogens |
| Fungi |
| Candida, Histoplasma, Cryptococcus, Aspergillus |
| Parasites |
| Toxoplasma, Toxocara, Cysticercus, Onchocerca |
| Noninfectious disorders |
| Autoimmune disorders |
| Behçet’s disease |
| Vogt–Koyanagi–Harada disease |
| Systemic lupus erythematosus |
| Wegener’s granulomatosis |
| Sympathetic ophthalmia |
| Retinal vasculitis |
| Malignancies |
| Intraocular lymphoma |
| Malignant melanoma |
| Leukemia |
| Metastatic lesions |
| Unknown etiology |
| Sarcoidosis |
| Serpiginous choroiditis |
| Acute multifocal placoid pigment epitheliopathy |
| Birdshot chorioretinopathy |
| Retinal pigment epitheliopathy |
| Multiple evanescent white dot syndrome |
Most cases of posterior uveitis are associated with some form of systemic disease. The cause can often be established on the basis of (1) the morphology of the lesions, (2) the mode of onset and course of the disease, or (3) the association with systemic symptoms or signs. Other considerations are the age of the patient and whether involvement is unilateral or bilateral. Laboratory and ancillary tests are often helpful.
Lesions of the posterior segment of the eye can be focal, multifocal, geographic, or diffuse. Those that tend to cause clouding of the overlying vitreous should be differentiated from those that give rise to little or no vitreous cells. The type and distribution of vitreous opacities should be described. Inflammatory lesions of the posterior segment are generally insidious in onset, but some may be accompanied by abrupt and profound visual loss.
Worldwide, the most common causes of retinitis in immunocompetent patients are toxoplasmosis, syphilis, and Behçet’s disease, whereas the most common causes of choroiditis are sarcoidosis, tuberculosis, and Vogt–Koyanagi–Harada disease. Inflammatory papillitis (optic neuritis) can be caused by any of these diseases, but multiple sclerosis should always be suspected, particularly when associated with eye pain worsened by movement (Chapter 14). Less common causes of posterior uveitis include intraocular lymphoma, acute retinal necrosis (ARN) syndrome, sympathetic ophthalmia, and the “white dot” syndromes such as multiple evanescent white dot syndrome (MEWDS) or acute multifocal posterior placoid epitheliopathy (AMPPE).
Diagnostic clues and clinical features of the more commonly encountered posterior uveitis syndromes are described below.
Posterior uveitis in patients under 3 years of age can be caused by a “masquerade syndrome” such as retinoblastoma or leukemia. Infectious causes of posterior uveitis in this age group include congenital toxoplasmosis, toxocariasis, and perinatal infections due to syphilis, cytomegalovirus, herpes simplex virus, varicella-zoster virus, or rubella virus.
In the age group from 4 to 15 years, the most common causes of posterior uveitis are toxoplasmosis and toxocariasis. Uncommon causes include syphilis, tuberculosis, sarcoidosis, Behçet’s syndrome, and Vogt–Koyanagi–Harada disease.
In the age group from 16 to 50 years, the differential diagnosis for posterior uveitis includes syphilis, tuberculosis, sarcoidosis, toxoplasmosis, Behçet’s disease, Vogt–Koyanagi–Harada disease, and ARN syndrome.
Patients over age 50 years who present with posterior uveitis may have syphilis, tuberculosis, sarcoidosis, intraocular lymphoma, birdshot chorioretinitis, ARN syndrome, toxoplasmosis, or endogenous endophthalmitis.
Unilateral posterior uveitis favors a diagnosis of toxoplasmosis, toxocariasis, ARN syndrome, or endogenous bacterial or fungal infection.
Reduced vision—Reduced visual acuity may be present in all types of posterior uveitis but especially in the setting of a macular lesion or retinal detachment. Every patient should be examined for an afferent pupillary defect, which, when present, signifies optic nerve or widespread retinal dysfunction.
Ocular injection—Eye redness is uncommon in strictly posterior uveitis but can be seen in diffuse uveitis.
Pain—Pain is atypical in posterior uveitis but can occur in endophthalmitis, posterior scleritis, or optic neuritis, particularly when caused by multiple sclerosis.
Signs important in the diagnosis of posterior uveitis include hypopyon formation, granuloma formation, glaucoma, vitritis, morphology of the lesions, vasculitis, retinal hemorrhages, and scar formation.
Hypopyon—Disorders of the posterior segment that may be associated with significant anterior inflammation and hypopyon include syphilis, tuberculosis, sarcoidosis, endogenous endophthalmitis, Behçet’s disease, and leptospirosis. When this occurs, the uveitis is more appropriately termed diffuse or panuveitis.
Type of uveitis—Anterior granulomatous uveitis may be associated with conditions that affect the posterior retina and choroid, including syphilis, tuberculosis, sarcoidosis, toxoplasmosis, Vogt–Koyanagi–Harada disease, and sympathetic ophthalmia. On the other hand, nongranulomatous anterior uveitis may be associated with Behçet’s disease, ARN syndrome, intraocular lymphoma, or the white dot syndromes.
Glaucoma—Acute ocular hypertension in association with posterior uveitis can occur with toxoplasmosis, ARN syndrome due to herpes simplex virus or varicella-zoster virus, sarcoidosis, or syphilis.
Vitritis—Posterior uveitis is often associated with vitritis, usually due to leakage from the inflammatory foci, from retinal vessels, or from the optic nerve head. Severe vitritis tends to occur with infections involving the posterior pole, such as toxoplasmic retinochoroiditis or bacterial endophthalmitis, whereas mild to moderate inflammation usually occurs with primary outer retinal and choroidal inflammatory disorders. Serpiginous choroiditis and presumed ocular histoplasmosis are typically accompanied by little if any vitritis.
Morphology and location of lesions—
Retina—The retina is the primary target of many types of infectious agents. Toxoplasmosis is the most common cause of retinitis in immunocompetent hosts. The active lesion of toxoplasmosis is generally seen in the company of old, healed scars that may be heavily pigmented. The lesions may appear in a juxtapapillary location and often give rise to retinal vasculitis. The vitreous is generally clouded when large lesions are present. In contrast, retinal infection with herpes viruses, such as cytomegalovirus and varicella-zoster virus, is more common in immunocompromised hosts. Rubella and rubeola virus retinal infections occur primarily in infants, where they tend to produce diffuse pigmentary changes involving the outer retina referred to as “salt and pepper” retinopathy (see Chapter 15).
Choroid—The choroid is the primary target of granulomatous processes such as tuberculosis and sarcoidosis. Patients with tuberculosis and sarcoidosis may present with a focal, multifocal, or geographic choroiditis. Both multifocal and diffuse infiltration of the choroid occur in Vogt–Koyanagi–Harada disease and sympathetic ophthalmia. Birdshot chorioretinopathy and presumed ocular histoplasmosis syndrome, in contrast, almost always produce multifocal choroiditis.
Optic nerve—Primary inflammatory optic neuritis can occur from syphilis, tuberculosis, sarcoidosis, toxoplasmosis, multiple sclerosis, Lyme disease, intraocular lymphoma, or systemic Bartonella henselae infection (cat-scratch disease). Peripapillary serous retinal detachment and/or macular star are often present.
A history of trauma in patients with uveitis raises the possibility of intraocular foreign body or sympathetic ophthalmia. Surgical trauma, including routine operations for cataract and glaucoma, may introduce micro-organisms into the eye and lead to acute or subacute endophthalmitis.
The onset of posterior uveitis may be acute and sudden or slow and insidious. Diseases of the posterior segment of the eye that tend to present with sudden loss of vision include toxoplasmic retinochoroiditis, ARN syndrome, and bacterial endophthalmitis. Most other causes of posterior uveitis have a more insidious onset.
Toxoplasmosis is caused by Toxoplasma gondii, an obligate intracellular protozoan. The ocular lesions may be acquired in utero or following systemic infection. Constitutional symptoms may be mild and easily missed. The domestic cat and other feline species serve as definitive hosts for the parasite. Susceptible women who acquire the disease during pregnancy may transmit the infection to the fetus, where it can be fatal. Sources of human infection include oocysts in soil or airborne in dust, undercooked meat containing bradyzoites (encysted forms of the parasite), and tachyzoites (proliferative form) transmitted across the placenta.
Patients with toxoplasmic retinochoroiditis present with a history of floaters and blurred vision. In severe cases there may also be pain and photophobia. The ocular lesions consist of fluffy-white areas of focal necrotic retinochoroiditis that may be small or large and single or multiple. Active edematous lesions are often adjacent to healed retinal scars (Figure 7–8). Retinal vasculitis and hemorrhage can be observed. Cystoid macular edema can accompany lesions in or near the macula. Iridocyclitis is frequently seen in patients with severe infections, and intraocular pressure may be elevated.
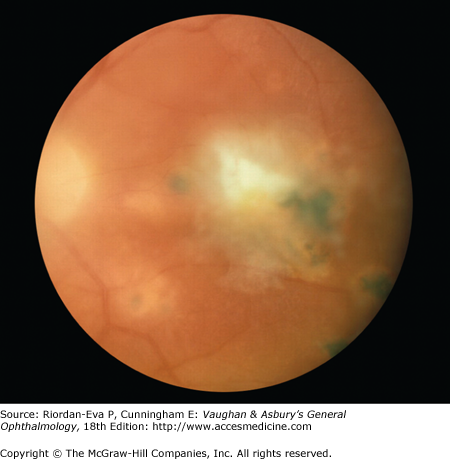
A positive serologic test for T gondii with consistent clinical signs is considered diagnostic. An increase in antibody titer is usually not detected during reactivation, but an elevated IgM titer provides strong evidence for recently acquired infection.
Small lesions in the retinal periphery not associated with significant vitritis require no treatment. In contrast, severe or posterior infections are usually treated for 4–6 weeks with pyrimethamine, 25–50 mg daily, and trisulfapyrimidine, 0.5–1 g four times daily. Loading doses of 75 mg of pyrimethamine daily for 2 days and 2 g of trisulfapyrimidine as a single dose should be given at the start of therapy. Patients are usually also given 3 mg of leucovorin calcium twice weekly to prevent bone marrow depression. A complete blood count should be performed weekly during therapy (Table 7–2).
An alternative approach for the treatment of ocular toxoplasmosis consists of administration of clindamycin, 300 mg four times daily, with trisulfapyrimidine, 0.5–1 g four times daily. Clindamycin causes pseudomembranous colitis in 10%–15% of patients. Other antibiotics effective in ocular toxoplasmosis include spiramycin and minocycline. Choroidal neovascularization can be treated with photodynamic therapy (PDT) or intra-vitreal anti-VEGF (vascular endothelial growth factor) injections.
Anterior uveitis associated with ocular toxoplasmosis may be treated with topical corticosteroids and cycloplegic/mydriatic agents. Periocular corticosteroid injections are contraindicated. Topical glaucoma medications are occasionally necessary. Systemic corticosteroids can be used in conjunction with antimicrobial therapy for vision-threatening inflammatory lesions but should never be used for a prolonged period in the absence of antimicrobial coverage.
In some areas of the United States where histoplasmosis is endemic (the Ohio and Mississippi River Valley areas), the diagnosis of choroiditis due to presumed ocular histoplasmosis is common. Patients usually have a positive skin test to histoplasmin and demonstrate “punched-out” spots in the posterior or peripheral fundus. These spots are small, irregularly round or oval, and usually depigmented centrally with a finely pigmented border. Peripapillary atrophy and hyperpigmentation occur frequently. Macular lesions may produce choroidal neovascularization, a complication that should be suspected in every patient with presumed ocular histoplasmosis who presents with decreased vision or evidence of subretinal fluid or hemorrhage. Choroidal neovascularization is effectively treated with corticosteroids, PDT or intra-vitreal anti-VEGF injections (see Chapter 10).
Toxocariasis results from infection with Toxocara cati (an intestinal parasite of cats) or Toxocara canis (an intestinal parasite of dogs). Visceral larva migrans is a disseminated systemic infection occurring in a young child (Table 7–5). Ocular involvement rarely occurs in visceral larva migrans.
| Visceral Larva Migrans | Ocular Larva Migrans1 | |
|---|---|---|
| Average age at onset | 2 years | 7 years |
| Fever | + | – |
| Abdominal symptoms (pain, nausea, diarrhea) | + | – |
| Nonspecific pulmonary disease | + | – |
| Hepatosplenomegaly | + | – |
| Eosinophilia | + | – |
| Hypergammaglobulinemia | + | – |
| ELISA (serum anti-Toxocara antibodies) | + | ± |
| ELISA (aqueous Toxocara antibodies) | – | + |
| Ocular finding1 | – | + |
Ocular toxocariasis may occur without systemic manifestations. Children acquire the disease by close association with pets and by eating dirt (pica) contaminated with toxocara ova. The ingested ova form larvae that penetrate the intestinal mucosa and gain access to the systemic circulation and finally to the eye. The parasite does not infect the intestinal tract of humans.
The disease is usually unilateral. Toxocara larvae lodge in the retina and die, leading to a marked inflammatory reaction and local production of toxocara antibodies. Children are typically brought to the ophthalmologist because of a red eye, blurred vision, or a whitish pupil (leukocoria).
Three clinical presentations are recognized: (1) a localized posterior granuloma, usually near the optic nerve head or fovea; (2) a peripheral granuloma involving the pars plana, often producing an elevated mass that mimics the snowbank of intermediate uveitis; and (3) chronic endophthalmitis.
Characteristic clinical findings and a positive enzyme-linked immunosorbent assay (ELISA) for anti-toxocara antibodies, even at low titer, confirm the diagnosis of ocular toxocariasis. Negative ELISAs are common but do not rule out the possibility of ocular infection. Positive antibody titers of the ocular fluids from patients with suspected ocular toxocariasis have been demonstrated in the setting of a negative serum ELISA, but the test is not routinely available and in any case is seldom necessary.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


