34 Tympanojugular Paragangliomas
Paragangliomas are tumors that arise from the paraganglionic system, aggregations of cells found throughout the body associated with vascular and neuronal adventitia.1,2 They originate from the neural crest and are related to the autonomic nervous system. The tumors of the head and neck are associated with the parasympathetic division, as opposed to the sympathetic division for the remaining parts of the body.3 There is, and has been a vast variance in the terminology for these tumors in the published literature. In truly histological terms a pheochromocytoma is a type of paraganglioma, and because of the relative rarity of extra-adrenal tumors of this type, the term pheochromocytoma has been often used to encompass all paraganglioma.2,4 The term chromaffin tumor relates to the characteristic histochemical reaction of cells that secrete catecholamines.1 Commonly used terminology describes a pheochromocytoma as a chromaffin tumor arising in the adrenal medulla, and paraganglioma or extra-adrenal pheochromocytomas for those arising in the sympathetic ganglia in the abdomen and chest.5 Nonchromaffin tumors (also previously called chemodectomas), correspond to paraganglioma arising from the parasympathetic division, that is, those of the head and neck.2 The term glomus tumor has long been used to describe paraganglioma of the head and neck. True glomus tumors or glomangiomas, however, are histologically separate cutaneous tumors, arising from neuromyoarterial cells around arteriovenous anastomoses.1,2,4
To simplify terminology the term pheochromocytoma should be used for those tumors arising in the adrenal, extra-adrenal abdominal, and thoracic locations to emphasize their endocrinologically active nature, while paraganglioma are those tumors arising in the head and neck and are nonsecreting in the vast majority.2,5,6 Therefore, the term glomus should be replaced with that of paraganglioma annotating its location, as outlined in the World Health Organization classification.3 Tympanojugular paragangliomas (TJPs) arise either in the adventitia of the jugular bulb or along the course of Jacobson nerve or Arnold nerve.1,4,7 The term tympanicum has been applied to those paragangliomas arising on the promontory and remaining confined to the middle ear and mastoid compartments, without erosion of the jugular plate and involvement of the jugular bulb.8 Jugular paragangliomas have been described as those arising from within the jugular bulb. The exact site of origin is often difficult to determine as paragangliomas can arise from within the canaliculi of the temporal bone, the jugular fossa and the middle ear cleft as they are in close proximity of each other.8
The vagal paragangliomas (VP) arise from the nodose ganglion in almost all cases.9 The carotid body tumors originate from the carotid body at the carotid bifurcation.10 Paragangliomas of the head and neck make up only 3% of all paragangliomas, comprising approximately 0.6% of head and neck tumors and 0.03% of all tumors.3 The overall incidence of head and neck paraganglioma (HNPG) ranges from 1 in 30,000 to 1 in 100,000, with carotid body paragangliomas making up nearly 60% of head and neck paraganglioma, TJPs nearly 40%, and vagal paraganglioma < 5%.3,6,7 The fact that carotid paragangliomas are far more common than other HNPG is probably because of a higher mass of normal paraganglionic tissue in this area.10,11 Carotid body tumors (CBT) and VP can be clinically grouped as cervico-carotid tumors.
TJPs constitute the second commonest tumor of the temporal bone and the commonest tumor affecting the jugular fossa.11 Tympanic paragangliomas are the commonest neoplasm affecting the middle ear. Therefore while rare, these are lesions frequently encountered by the skull base and head and neck surgeon. Paragangliomas can arise as both sporadic and familial entities, a fact first documented in 1933 by Chase, in a description of bilateral CBT existing in sisters.12
It is now known that 25 to 35% of paragangliomas are associated with recognized genetic defects, most of which are because of hereditary transmission.6,7 These defects are usually associated with one of the four familial paraganglioma syndromes. Mutations of SDHD are linked to paraganglioma locus 1 (PGL 1), SDHC with PGL 3, and SDHB with PGL 4.6,7 The genetic defect in PGL 2 is still unclear. This means that approximately 30% of apparently sporadic HNPG are because of one of these defects. Multiple tumors are not uncommon, found in 10 to 20% of sporadic cases, and in up to 80% of familial cases.12,13 All subtypes of HNPG show a peak age of presentation in the fourth and fifth decades, with rare incidences of pediatric cases.13 While the sex ratio is equal in carotid paraganglioma, females are affected 4 to 6 times more than males in TJPs. Males, however, are more commonly affected in the familial type. Those with a familial etiology, however, present in the second decade.13–15
High altitude appears to increase the incidence of CBT but not in any other head and neck paraganglioma, with a significant female preponderance.16 There are also geographical variances in the incidence of head and neck paragangliomas, because of founder effects of genetic mutations. This is most markedly seen in the Netherlands.17
Pathology and Genetics
Structurally, temporal bone paraganglia are 1 to 1.5 mm in length. Approximately half of the tympanojugular paraganglia are found in the adventitia of the anterolateral region of the jugular bulb, in close proximity to Jacobson nerve, within the inferior tympanic canaliculus or over the cochlear promontory.18,19 The remaining paraganglia are found along the course of the Arnold nerve in the mastoid canaliculus. Very rarely they can be found associated with the connection between Arnold nerve and the vertical segment of the facial nerve (FN), or within the fallopian canal itself.20 This distribution of paraganglia thus accounts for the site of origin of paragangliomas of the temporal bone.21
VP are associated with the epineurium of the cranial nerve X, and are most commonly associated with the inferior or nodose ganglion.22 All paragangliomas have neurosecretory granules, but only 0 to 4% of HNPG are functional.23 Paragangliomas are predominantly benign, slow growing, highly vascular tumors, but they have propensity for aggressive local destruction. Because of this clinical behavior they have the ability to cause significant morbidity, especially when arising in relationship to the skull base with its multitude of associated neurovascular structures.
Because of their generally slow growth and initial absence of symptoms, jugular paragangliomas are often not detected until of a significant size. Those arising initially in the tympanic cavity, however, are usually detected at an earlier stage because of the presentation with hearing loss from ossicular chain interference, and or pulsatile tinnitus.24 With progression, jugular paraganglioma most frequently follow a path of least resistance into the middle ear cleft and within the jugular vein.24 Further spread then occurs through air cell tracts to involve the intrapetrous carotid canal, along the eustachian tube, into the neck along the carotid sheath, and, in later stages, intracranially. Tumor can also extend along the inferior petrosal sinus. Intracranial spread usually occurs through the medial wall of the jugular fossa.24 Lower cranial nerve involvement occurs later and is usually related to invasion through the medial wall of the jugular bulb. The FN lies in close proximity to the jugular bulb in its vertical segment and is also at risk.24 The pattern of tumor extension is accompanied by an often underestimated degree of bony infiltration. The tympanic bone is widely involved early, with extension in all directions. This characteristic bony erosion is related to ischemic necrosis. The otic capsule is relatively spared but extension to the inner ear can certainly occur. Invasion of the jugular tubercle, occipital condyle and subsequently the cranial nerve XII is because of medial extension with further postero-inferior spread leading to vertebral artery involvement.24 Further anteromedial spread can involve the cavernous sinus, with significant intra-dural involvement usually found in the advanced stages of disease.25
There is little doubt that paragangliomas as a group display a slow growth rate. Jansen et al studied a mixed group of HNPG by a wait and scan policy. Sixty percent exhibited growth, and of these the median tumor doubling time was calculated to be 4.2 years. Seventy-nine percent of their patients had multiple tumors and they found very small and very large tumors to have the slowest growth rates.26,27 Unfortunately no comment was made regarding the age or stage of these lesions. There appears to be however a subgroup of tumors that display a much more aggressive natural history. This represents approximately 15 to 30% of TJPs.28 It is tumors arising in younger patients that tend to behave in a more aggressive fashion, with advanced disease at presentation and which are responsible for higher recurrence rates following treatment.29 There continues to be some variance in the literature regarding the nature of paraganglioma, which are at odds with the fact that there is a significant degree of infiltration of the bone of the skull base.30 The degree of cranial nerve infiltration is also a point of debate. The medial wall of the jugular bulb represents a barrier between tumor and nerves; however once this barrier has been breached envelopment and/or infiltration of the nerves is the rule.29,30
Approximately 2 to 5% of TJPs are found to be malignant.31 This compares to VP with a probable rate of malignancy of 10%, but reported to be as high as 19%, and carotid body paraganglioma that have a rate from 1.4 to 12%.32,33 Not dissimilar to the clinical behavior of the primary tumor, many metastatic deposits display indolent characteristics. However as a group, metastatic tumors are more aggressive, with higher rates of recurrent disease, and ultimately disease related mortality.34 Regional lymph nodes are the commonest site for metastases. Less frequently bone, lung, and liver can be sites of metastatic disease.35 Lee et al reported that approximately 70% of metastases were confined to regional lymph nodes at presentation, and in the case of carotid body tumors confinement occurs in 94%.36 To identify malignant tumors it is important to sample lymph nodes. The lymph nodes that should be sampled during surgery are, for cervico-carotid tumors: levels II and III lymph nodes and for TJPs: level II lymph nodes.37
Clinical Presentation
The majority of cases initially present with pulsatile tinnitus with other symptoms as a consequence of the specific growth pattern of the tumor.38 Hearing loss can occur if the middle ear and ossicles are affected and lower cranial nerve deficits can develop with invasion of the jugular foramen.24,39 These symptoms develop slowly and these patients often are able to compensate for the progressive loss of lower cranial nerve functions. In the majority of cases a red mass is visible on otoscopic examination. It can be a retrotympanic mass for lesions expanding into the middle ear cleft, while tumors invading the tympanic bone from below can show the classic “rising sun” sign.40 These tumors can also extend through the tympanic membrane and can be confused with an inflammatory polyp. Any vascular mass seen on otoscopy, with unclear margins, involves the jugular bulb until proven otherwise.24,28 All patients will require a full cranial nerve examination including upper aerodigestive tract endoscopy and careful neck examination. Routine screening for urinary catecholamines is recommended despite the low rates of secreting tumors.41
Diagnosis
Computed tomography (CT) and magnetic resonance imaging (MRI) are mandatory and complementary in the evaluation of any jugular foramen lesion. In fact while MRI with gadolinium allows a better understanding of the tumor extension as well as neck and intracranial involvement, CT permits better assessment of bony erosion.24,25,42 Low to intermediate T1 signal and relatively high T2 signal are typical for paragangliomas.24 A classic “salt and pepper” pattern in TJPs > 2 cm is seen on T1 after gadolinium infusion, because of the presence of intratumoral vasculature appearing as flow voids.28,29,43 Dural invasion is not always easy to detect, because often the dura is infiltrated and pushed medially, without true invasion of the posterior fossa. Sagittal sections allow to appreciate the intra- and extracranial extension of the lesion (Fig. 34.1).33,34,44 Extension through the jugular plate from either direction occurs early, with involvement of the hypotympanum and jugular bulb. Early changes are represented as an indistinct lateral margin of the jugular foramen, followed by erosion of the caroticojugular crest and jugular spine (Fig. 34.2).24,28,44 The extent of involvement of the infralabyrinthine cell tract and carotid canal is used to classify the lesion according to the Fisch classification. As mentioned, the degree of bony involvement is often difficult to assess. This is especially problematic in upstaging early lesions, necessitating the conversion of a transmastoid to an infratemporal type A approach, as well as in extensive involvement of the petrous apex, clivus anteriorly, and occipital condyle and hyoglossal canal posteriorly.24,28,45,46
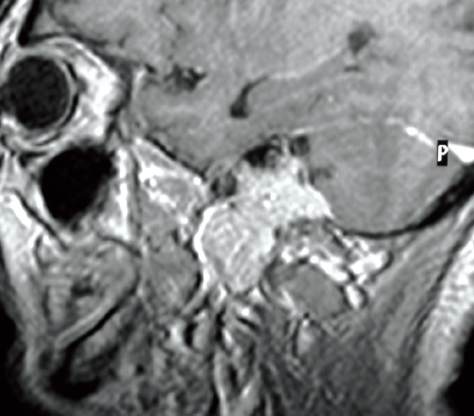
Figure 34.1 Sagittal magnetic resonance imaging showing intracranial extension.
©2010 Thieme Publishers, Stuttgart, Germany. Printed with permission.
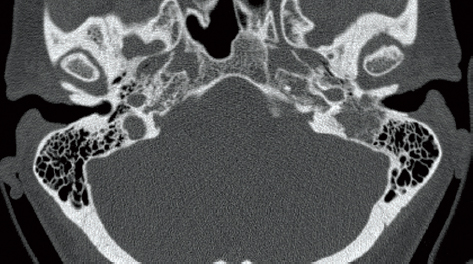
Figure 34.2 Characteristic erosion of the temporal bone noted on computed tomography scan.
©2010 Thieme Publishers, Stuttgart, Germany. Printed with permission.
Two classification systems are in common use, that of Fisch and that of Glasscock-Jackson.21 In terms of describing the involvement of the internal carotid artery (ICA), the most critical aspect in planning the surgical approach, it is Fisch system that we recommend be used. There is also a close correlation between the C class and the likelihood of intracranial extension.
In the majority of cases CT and MRI allow a clear differentiation from other lesions involving the jugular foramen area, mainly jugular foramen schwannomas and meningiomas.24 Schwannomas characteristically reveal smooth enlargement of the jugular foramen, with early sparing of the lateral wall. While meningiomas can produce an irregular margin of the jugular foramen, the degree of bony erosion is significantly less and are often associated with hyperostosis.46 Meningiomas may also show intratumoral calcification and a dural tail while intratumoral cysts are more frequent in schwannomas.47
The current use of magnetic resonance angiography and venous phase CT normally provides adequate information regarding the arterial and venous circulation status.
Four vessel digital angiography with embolization is integral in the management of TJP.24,28 It can also aid in the diagnosis in difficult cases. TJPs show a coarse blush, greater than that of a meningioma, and rapid diffusion on venous phase because of rapidly draining veins.24 The primary blood supply is from the ascending pharyngeal artery, gaining additional supply from both external and internal carotid systems with tumor growth (Fig. 34.3).24 Screening for coexistent lesions is also very important. MRI scanning of the entire neck is required, with investigation of the abdomen if there are any signs of catecholamine excess or family history.
Treatment Options and Decision Making
In general, the options for management of paragangliomas include surgery, radiotherapy, or a follow-up policy. In the majority of these lesions, the surgery remains the mainstay of treatment. Total removal leads to complete cure in most cases. The complex anatomy and the close relation to vital structure mandate a wide exposure to facilitate tumor removal without endangering these related structures. The hallmark of skull base surgery is to remove the bone to provide the necessary access to remove the tumor.
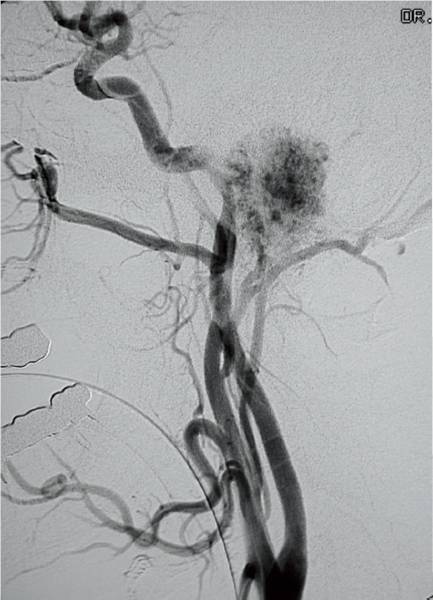
Figure 34.3 Angiography showing the characteristic tumor blush seen with tympanojugular paragangliomas.
©2010 Thieme Publishers, Stuttgart, Germany. Printed with permission.
• The ideal approach would achieve the following24:
• Wide access to the whole tumor permitting total removal.
• Minimized morbidity and mortality.
• No additional deficit related to the approach.
• Access should be near the tumor, so that the surgeon is working at a comfortable angle.
• Ease and rapidity of performance
As the tumor class and size are the most important parameters that determines the management plan, tumors will be grouped under different classes according to the most difficult and challenging sites and decision making will be discussed accordingly (Tables 34.1 and 34.2).
Solitary Lesions
The goal of treatment can be either one of cure, control or of palliation. This is reflected in the general management options24:
• Total surgical removal.
• Planned subtotal removal aiming to preserve neurovascular structures with or without postoperative radiotherapy.
• Partial resection for symptomatic control.
• Primary radiotherapy.
• Conservative or wait and scan policy.
Often patient factors are just as important as the extent of pathology when formulating a management plan. The following parameters should be carefully taken into consideration24,46:
• Patient’s age, general medical condition, life expectancy
• Lower cranial nerve function, mainly the vagus nerve
• Tumor size, with particular regard to intracranial extension and carotid artery involvement
• Collateral venous drainage: significantly the patency of the contralateral sigmoid sinus and jugular bulb
• FN function
• Hearing status
It is our strong belief that microsurgery remains the preferred treatment modality in the majority of cases, especially in patients with a life expectancy of longer than 30 years.
Tympanic and Tympanomastoid Paragangliomas
There is no benefit to be gained by delaying definitive surgical treatment. Recurrence rates and additional morbidity related to the intervention are very low. There is additionally no role for radiotherapy in these lesions.24 However, elderly patients affected by class B2 or B3 tumors may be enlisted in a wait and scan policy, because the surgery may not be able to restore the hearing and that is often the only deficit induced by the lesion.26 Because of the common slow growth of the tumors, their enlargement to a dangerous size is extremely rare in a short time span. For the same reasons subtotal removal may be another option to keep into consideration in this group of patients. Based on our experience we have introduced formulated a surgical strategy in accordance to the following classification (Table 34.1, Figs. 34.4 and 34.5).24
Tympanojugular Paragangliomas
Because of their unique location, these tumors pose certain problems (Table 34.2):
• Injury of the lower cranial nerves.
Table 34.1 Modified Fisch Classification of Tympanic and Tympanomastoid Paragangliomas
Class A | Tumors limited to the middle ear cleft without invasion of the hypotympanum |
A1 | Tumors completely visible on otoscopic examination. |
A2 | Tumor margins are not visible on otoscopy. Tumor may extend anteriorly to the eustachian tube and/or to the posterior mesotympanum. |
Class B | Tumors limited to the tympanomastoid compartment of the temporal bone without erosion of the jugular bulb |
B1 | Tumors confined to the middle ear cleft with extension to the hypotympanum. |
B2 | Tumors involving the middle ear cleft with extension to the hypotympanum and the mastoid. |
B3 | Tumors confined to the tympanomastoid compartment with erosion of the carotid canal. |
• The FN is centered on and is closely related to the jugular bulb.
• These tumors spread to four different “compartments”: medially intradural, anteriorly intrapetrous, inferiorly extracranial to the neck, and posteroinferiorly toward the condyle and vertebral artery.
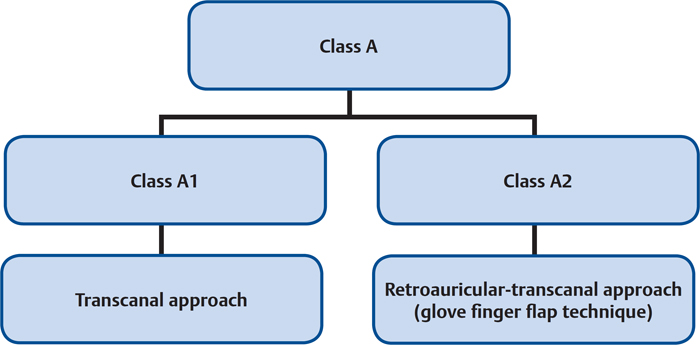
Figure 34.4 Algorithm for the surgical management of the various types of class A tumors.
©2010 Thieme Publishers, Stuttgart, Germany. Printed with permission.
Young patients with impairment of the vagus nerve should be treated surgically. It must be acknowledged however that further lower cranial nerve injury is likely.
Compensation following acute compound lower cranial nerve palsies is particularly difficult in elderly patient and normal lower cranial nerve function should be considered a relative contraindication for surgery in patients older than 60 years.47 The same is true for patient with coexisting respiratory problems. Despite our usual approach to achieve surgical cure, in the situation of advanced age or poor medical status there is rarely an indication for radical surgical removal. Radiological follow-up often represents the best treatment in these cases. Radiotherapy is reserved for patients who document significant growth on follow-up.31
Surgical removal of TJP implies sacrifice of the jugular bulb. Usually the venous pathway is already occluded by the tumor and it may be resected without any consequence. However, in particular cases the bulb is still patent or the compensation has occurred through collaterals, such as the posterior condylar vein, that have to be sacrificed as well.48 When this happens in the presence of a hypoplasia of the contralateral venous system sacrifice of the bulb means occlusion of the main venous drainage of the brain, with the consequent risk of benign intracranial hypertension or venous infarction of the temporal lobe.48 In this situation it is advisable to wait till the occlusion of the bulb by the tumor growth before planning the surgery.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


