Fig. 1
Normal trachea with rigid cartilaginous support
Other abnormalities associated with tracheomalacia include a variety of syndromes and additional aerodigestive, cardiac, and neurologic anomalies (Table 1). In their series, Masters et al. found that tracheomalacia was associated with tracheoesophageal fistula (TEF) in 9.7 %. Eight percent of patients had defined syndromes [35]. Studies have found cardiovascular anomalies in 14–58 % of patients with TM, including patent ductus arteriosus, atrial or ventricular septal defects, vascular ring anomalies such as double aortic arch or pulmonary artery sling, hypoplastic left or right heart, Tetralogy of Fallot and other conotruncal cardiac disorders (affecting the cardiac outflow tract), dextrocardia, and valvular stenosis [10, 33, 35]. Bronchopulmonary dysplasia may be seen in up to 52 % of infants with TM, and gastroesophageal reflux disease is seen in about half of such patients. Subglottic stenosis, laryngomalacia, vocal cord paralysis, neurologic impairment, and overall developmental delay may also occur in association with TM [16]. Interestingly, 24 % of patients with laryngomalacia, a much more common condition, have also been found to have more distal airway malacia involving the trachea and/or mainstem bronchi [35].
Congenital (primary) | Acquired (secondary) |
|---|---|
Normal infants (idiopathic) | Prolonged intubation |
Prematurity | Tracheotomy |
Pulsatile collapse with normal innominate artery | Severe tracheobronchitis |
Congenital cartilage abnormalities | Compressive |
Dyschondroplasia, chondromalacia, chondrodysplasia, polychondritis, Ehlers–Danlos syndrome | Vascular, cardiac, skeletal, tumors and cysts, infection, post-traumatic |
Congenital syndromes associated with TBM | |
Mucopolysaccharidosis (Hurler syndrome, Hunter syndrome), CHARGE syndrome, VATER anomaly, trisomy 9, trisomy 21, 22q11 deletion, DiGeorge syndrome, Pierre-Robin sequence, Pfieffer syndrome, Larsen syndrome | |
Congenital anomalies associated with TBM | |
Tracheoesophageal fistula, esophageal atresia with/without laryngeal cleft, bronchopulmonary dysplasia |
Pathogenesis
In order to fully understand the clinical presentation of a patient with tracheobronchomalacia, a brief review of the physical mechanics of the respiratory cycle is in order. In isolation, the natural tendency of the chest wall is to expand. Conversely, the natural tendency of the lungs is to collapse. In order to maintain inflation of the lungs, intrathoracic pressure must therefore be negative. With inspiration, the diaphragm contracts and descends and the chest wall expands, increasing thoracic volume and making intrathoracic pressure more negative. The lungs expand, and a gradient for airflow into the lungs is established. With expiration, the opposite occurs, and the lungs deflate until the gradient for airflow no longer exists.
Key to this model of normal pulmonary dynamics is that the trachea and bronchi maintain their shape in the face of fluctuating intrathoracic pressures. During inspiration, both intrathoracic pressure and intraluminal airway pressure are negative. During expiration, however, intrathoracic pressure becomes positive in relation to intraluminal airway pressure, and the cartilaginous rings of the normal trachea and bronchi serve to provide the necessary rigidity to resist the external force and prevent airway collapse (Fig. 2). If these cartilaginous elements fail to provide such support, as in tracheobronchomalacia, the airway collapses and obstruction occurs (Fig. 3).
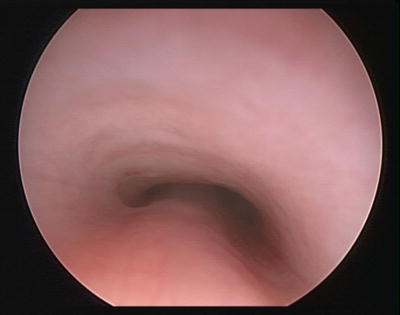
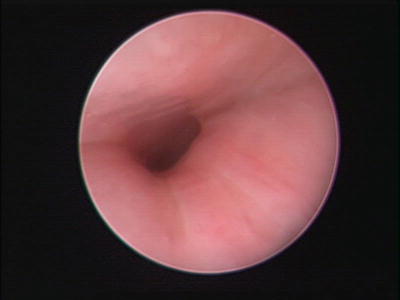
Fig. 2
Primary tracheomalacia with soft, collapsible cartilaginous support proximal to the carina
Fig. 3
Isolated left mainstem bronchomalacia
Baxter and Dunbar were the first to divide tracheomalacia into primary and secondary forms. Primary airway malacia was subsequently defined by Benjamin as isolated airway malacia in otherwise normal infants. Secondary airway malacia was defined as acquired malacia associated with TEF, esophageal atresia (EA), VATER/VACTERL association, vascular or external compression of the airways, or specific syndromes [6]. Disease processes that result in the formation of an abnormal cartilaginous matrix such as polychondritis and chondromalacia have since been included as primary forms, as have mucopolysaccharidoses such as Hunter syndrome and Hurler syndrome and other genetic syndromes [16].
Some controversy exists over the grouping of TM associated with TEF (Fig. 4) as an acquired rather than a congenital form of disease as there is inherent weakness of the involved portion of the trachea [16]. In 75 % of infants with TEF and EA, the trachea is more easily collapsible due to reduced circumference of tracheal cartilage and widening of the membranous trachea [50]. Blair and colleagues identified esophageal muscle in the histologic specimens taken from the posterior wall of the trachea in infants with TEF and/or EA. They subsequently proposed that idiopathic primary TM is actually the result of a defect in foregut division resulting in inherent tracheal weakness, just not severe enough to cause TEF or EA [10]. Others have supported the theory that congenital TM is due to faulty embryologic division of the foregut into the trachea and esophagus, suggesting that the trachea receives redundant tissue during separation with a subsequent decrease in structural integrity [33]. As a result of such findings, current classification schemes class TM associated with TEF under a primary, or congenital, form of disease (Table 1).
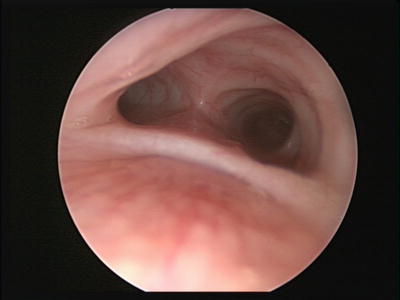
Fig. 4
Tracheomalacia in association with tracheoesophageal fistula
With such debate, Mair and Parsons were careful to reserve the term “tracheomalacia” for specific cases in which the posterior membranous wall of the trachea is widened. They noted a normal trachea to have a ratio of cartilage to posterior membrane of approximately 4–5:1, while in tracheomalacia this ratio approaches 2:1 [33]. In order to accommodate the entire spectrum of disease, they preferred to use the more encompassing term “major airway collapse” (MAC). They proposed a MAC classification system that includes three types. MAC Type I lesions represent congenital tracheal collapse due to abnormal cartilaginous development, including that seen in premature infants and those with EA, TEF, mucopolysaccharidoses, and Larsen syndrome. MAC type II lesions consist of tracheal collapse due to extrinsic compression alone. MAC type III lesions are acquired lesions, generally due to inflammatory stimuli such as severe or recurrent tracheobronchitis, gastroesophageal reflux disease, and intubation [33, 42].
Acquired TM is a result of the degeneration of normal cartilaginous support from a variety of causes and is slightly more common than congenital disease. MAC type III inflammatory lesions are the most common acquired lesion and are most often associated with prolonged endotracheal intubation with associated increases in airway pressure, oxygen toxicity, and recurrent infections [52]. Premature infants with respiratory distress syndrome are most commonly predisposed to the condition. One study of 50 infants with TM found that 52 % had “secondary” TM, and 96 % of these were premature infants with respiratory distress syndrome and a history of prolonged mechanical ventilation [30]. It should be noted, however, that it could be difficult to differentiate whether TM is truly a result of endotracheal intubation or a contributing factor in the need for endotracheal intubation from the outset [20, 30]. Tracheotomy also predisposes to TM, so a tracheotomy performed to help in management of primary TBM at one site may predispose to secondary tracheomalacia at the surgical site. Tracheotomy requires surgical interruption of the integrity of the tracheal rings, and over time the tube’s presence may lead to cellular hypoxia and pressure necrosis due to impaired blood supply, tracheitis, and frictional mucosal damage [4, 22]. Finally, the role of gastroesophageal reflux as an inflammatory stimulus in the exacerbation and/or development of tracheomalacia may also be significant as it is more prevalent in children with large-airway malacia [9].
MAC type II lesions due to external tracheal compression present a less common form of acquired TM. Compression affects the integrity of the tracheal wall, at times severe enough to cause chronic obstruction but often only leading to transient collapse as intrathoracic pressure increases. Implicated structures and anomalies include double aortic arch (Fig. 5), abnormal branching of the innominate artery (Fig. 6), anomalous left pulmonary artery, right aortic arch with left ligamentum arteriosum, left atrial hypertrophy, and other vascular rings. Skeletal disorders such as scoliosis and pectus excavatum may contribute as well. Mass lesions such as thymus enlargement, hemangiomas, lymphatic malformations, teratomas, other tumors, abscesses, cysts, and thyroid goiters are also implicated. Importantly, even after surgical relief of the compression, tracheal weakness and collapse may persist and require additional treatment [16].
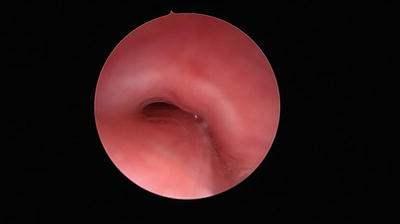
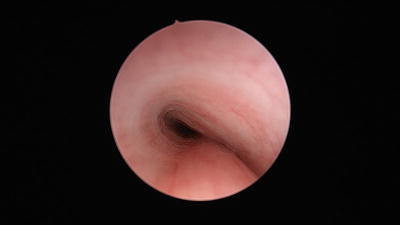
Fig. 5
Tracheomalacia in association with a double aortic arch
Fig. 6
Tracheomalacia in association with innominate artery compression and a large thymus
Clinical Presentation
Since it was first defined in the 1950s and 1960s, the signs and symptoms of TBM have been classically described as appearing slowly over the first several weeks of life. A more recent study published in 1992, however, found that symptoms of primary TBM are apparent at birth in up to 95 % of infants [33]. Mild symptoms may include noisy breathing, intermittent stridor, and wheezing. More severe signs and symptoms include severe dyspnea, spontaneous hyperextension of the neck, increased work of breathing with retractions, cyanosis, and respiratory insufficiency or failure. Symptoms are often positionally dependent, better when prone and worse when supine. Generally, severe cases are detected in the neonatal period when patients present with acute episodes of severe obstruction, cyanosis, and desaturation and at times ventilator dependency. Recurrent lower airway infections and pneumonia may result from impaired mucociliary function as well as impaired clearance of secretions by the collapsed and obstructed airway lumen during cough [44].
The most common symptoms seen in isolated tracheobronchomalacia are expiratory stridor and a “brassy” or “barky” cough. Negative intraluminal pressure compared to intrathoracic pressure causes dynamic collapse of the insufficiently supportive cartilaginous elements of the airway. Airway obstruction can occur at any phase of respiration but is most pronounced with expiration, and resultant airway turbulence causes the high-pitched sound of stridor. The majority of the trachea and the entirety of the bronchi lie within the thoracic cavity and respond to changes in pressure accordingly.
While expiratory stridor is the most common presentation of tracheomalacia, it may be confused with the wheezing found in reactive airway disease or bronchiolitis. In all cases airway noise may worsen with activity or agitation as airflow increases. Similarly, it will generally worsen with upper respiratory tract infections as an already compromised airway becomes narrowed by edema and thicker, more abundant secretions. In contrast to the high-pitched and varied “squeaking” heard with the wheezing of reactive airway disease, the stridor associated with tracheomalacia is more centrally located and of a more uniform, slightly lower pitch. In addition, symptoms generally lessen when the patient is prone as the gravitational pressure of the intrathoracic structures on the collapsible airway present when supine is reversed. Symptoms that fail to improve when the patient is at rest or with positioning maneuvers should raise suspicion of fixed intraluminal airway obstruction or possible extrinsic airway compression.
Nevertheless, the classic presentation of tracheobronchomalacia as purely expiratory stridor may not always be so straightforward. A portion of the trachea is cervical and not subject to intrathoracic pressure. In addition, the underlying cartilaginous immaturity found in tracheomalacia is not uncommonly present in the larynx as well, causing laryngomalacia. Without the effect of negative intrathoracic pressure, these cervical structures are subject only to Bernoulli’s principle. Briefly, an increase in airflow, i.e. during inspiration, causes a decrease in airway pressure. Intraluminal pressure becomes negative in relation to atmospheric pressure. Clinically insufficient airway support then allows collapse of the cervical airway and may cause inspiratory airway collapse and inspiratory stridor.
Though respiratory symptoms are generally the most alarming, feeding difficulties should not be overlooked. Dysphagia, regurgitation, cough, and cyanosis during feeding are not uncommon, particularly in association with vascular tracheal compression. During feeding, as the esophagus becomes engorged it may contribute to tracheal collapse and obstruction. Respiratory obstruction and desaturation may result, inhibiting the ability of the infant to feed normally and gain weight appropriately. Frequent interruption of feeding has been promoted as a means of preventing significant desaturation and cyanosis [10].
Finally, while the focus of this chapter is tracheobronchomalacia during the neonatal period, it should be noted that children with mild airway malacia may not present until after the neonatal period and will often have nonspecific symptoms such as rattling, wheeze, stridor, exercise intolerance, cough, and recurrent lower airway infections. In such patients it may be misdiagnosed as asthma, and due to its expected lack of response to standard asthma treatment may be further misdiagnosed as severe-persistent or therapy-resistant asthma [12].
Diagnosis
A thorough history and physical examination are critical as diagnosis is initially based upon the clinical presentation outlined above. As noted, TBM must be differentiated from asthma and other causes of intraluminal obstruction, and a high degree of suspicion is often necessary for accurate diagnosis. Reviews have demonstrated a mean delay from symptom onset to accurate diagnosis of 6–9 months [33, 47]. In mild cases with benign, intermittent noisy breathing that resolves at rest and with prone positioning and no evidence of clinically significant obstruction or increased work of breathing, clinical diagnosis may be sufficient. In cases where the diagnosis is in question, or when clinically apparent respiratory obstruction, increased work of breathing, and/or oxygen desaturation occurs, further diagnostic studies should be pursued.
The gold standard of diagnosis is operative bronchoscopy by an experienced airway endoscopist and anesthesia team. Communication between the surgeon and anesthesiologist is critical, as spontaneous ventilation is vital to achieving an accurate diagnosis. Only when the patient is breathing spontaneously, i.e. without positive-pressure support from the ventilator, can the dynamic airway collapse of tracheobronchomalacia be clearly seen. Therefore general inhalational anesthesia is necessary to avoid paralysis, heavy sedation, and positive-pressure ventilation that may impair recognition of dynamic airway collapse.
Based upon studies conducted in the 1960s by Wittenborg et al., a visualized decrease in airway diameter of >50 % is considered abnormal [54]. Most infants with TM will have >75 % collapse with complete collapse in up to 33 % of infants [54]. Collapse typically occurs in an anterior–posterior direction. Vascular compression may be appreciated with pulsatile collapse, and the level of collapse may help predict which vessel is likely to be the source of compression. A high riding innominate artery causes pulsatile compression of the anterior trachea a few centimeters above the carina, and compression of the mass with the bronchoscope will result in loss of the right upper extremity pulses. A double aortic arch, as the only complete vascular ring, will cause more pronounced pulsatile compression than other rings such as a right aortic arch with a left ligamentum arteriosum and aberrant left subclavian artery [46]. A pulmonary artery sling is classically associated with a “saber-shaped” tracheal stenosis and right middle segment bronchomalacia as the left pulmonary artery arises off of the right pulmonary artery and encircles the right mainstem bronchus and trachea as it courses between the trachea and esophagus towards the left lung [35].
Rigid bronchoscopy offers superior optics and, therefore better assessment of the tracheal mucosa and structure, with tracheal rings often distinct on visual inspection. It may be limited in its ability to assess past the level of the mainstem bronchi, however. Flexible bronchoscopy provides inferior image detail but may provide a more accurate picture of dynamic airway movement as less laryngeal manipulation is required to access the airway. Flexible bronchoscopy is also useful for assessing the distal bronchi for malacic change.
Tracheobronchomalacia’s association with congenital heart lesions, especially conotruncal lesions, warrants consideration of a full cardiac evaluation when a diagnosis is made. Conotruncal heart defects as well as vascular rings have been described in association with 22q11 chromosomal defects such as DiGeorge syndrome and velocardiofacial syndrome, so providers should consider genetic and immunologic assessment for associated defects [35].
Nonoperative investigation is often invaluable in the evaluation and diagnosis of TBM and can be divided into static imaging, dynamic imaging, and spirometry. Static imaging includes plain films of the chest, chest CT, and chest MRI. Dynamic imaging generally includes airway fluoroscopy and esophagram, though development continues in using dynamic MRI and cine-CT as dynamic modalities. Diagnostic chest radiographs, as the most basic form of static imaging, include inspiratory and expiratory views. Tracheal narrowing may be seen on an expiratory film, and lung fields may demonstrate parenchymal abnormalities consistent with airway collapse and consolidation [35]. However, when compared to microlaryngoscopy and bronchoscopy, the sensitivity of plain films in diagnosing TM is only 62 % [51].
Airway fluoroscopy has been utilized in the evaluation of suspected TBM. Strauss et al. reported airway fluoroscopy to be as good as or even superior to bronchoscopy in the diagnosis of patients with combined stenosis and tracheomalacia [48]. A more recent study compared airway fluoroscopy to the gold standard of operative bronchoscopy and confirmed a specificity of 94 % for a diagnosis of tracheomalacia achieved by airway fluoroscopy. However, its sensitivity was only 20 % in detecting tracheomalacia, so its role as a screening study may be limited and further investigation is certainly warranted if clinical suspicion exists [7]. Barium esophagography may be useful when concern for associated TEF, EA, or reflux exists. It may also demonstrate esophageal compression due to a double aortic arch.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


