12 Thyroid-Stimulating Hormone Tumors
 Epidemiology
Epidemiology
As early as the 1950s it was postulated that some forms of hyperthyroidism could have their origins in the pituitary gland.1 However, it was not until the 1960s that the first cases of thyroid-stimulating hormone (TSH)—secreting tumors were reported by Jailer and Holub.2 With the advent of newer immunostaining techniques, the incidence of TSH adenomas is reported as 2%.3 However, the incidence varies among series and is commonly reported to be approximately 0.5 to 3%.4,5
It is reported that TSH adenomas are more common in females.4,6 The female-to-male ratio varies widely in the literature from between 5:1 and 10:17 to 1:1.6.8 It has been suggested that males are diagnosed earlier than females due to the rarity of Graves’ disease in males, thus prompting early investigation for secondary hyperthyroidism.6 The age of diagnosis tends to be in middle adulthood, usually in the fifth or sixth decade. Though uncommon, TSH adenomas have been reported in children as young as 15 and in adults older than 80 years.6 Some studies have found that women tend to develop their adenomas at a younger age, have a longer history of symptoms, and have smaller tumors that are less invasive when compared with these tumors in men.4
 Pathology
Pathology
Histopathology
Thyrotrophic adenoma cells usually appear chromophobic with mild periodic acid-Schiff positivity. Immunostaining is positive for TSH-β in virtually all adenomatous cells from TSH adenomas (Fig. 12.1). Under electron microscopy the cells demonstrate moderate differentiation, scant rough endoplasmic reticulum (RER), and Golgi complexes. Secretory granules are small, round, evenly electron dense and are found along the cytoplasmic membrane9 (Fig. 12.2). Specialized techniques have identified the presence of TSH and other pituitary hormones within the same adenomatous cell and even from within the same secretory granule. Pituitary adenomas of the growth hormone (GH), prolactin (PRL), and TSH lineage may originate from a common progenitor cell; however, the presence of positive immuno-histochemistry does not necessarily mean that the patient will have hypersecretion of that hormone. It has been suggested that the production of two seemingly unrelated hormones by the same adenomatous cell is due to TSH adenomas developing from this common progenitor cell.
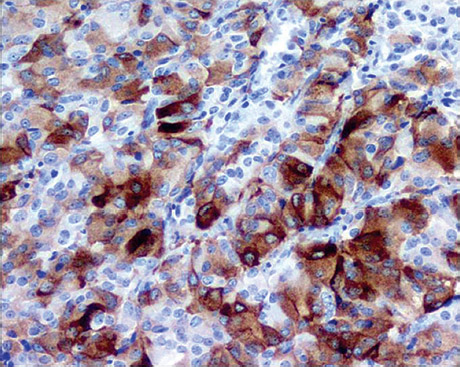
Fig. 12.1 Immunohistochemistry specimen demonstrating thyroid-stimulating hormone positivity.
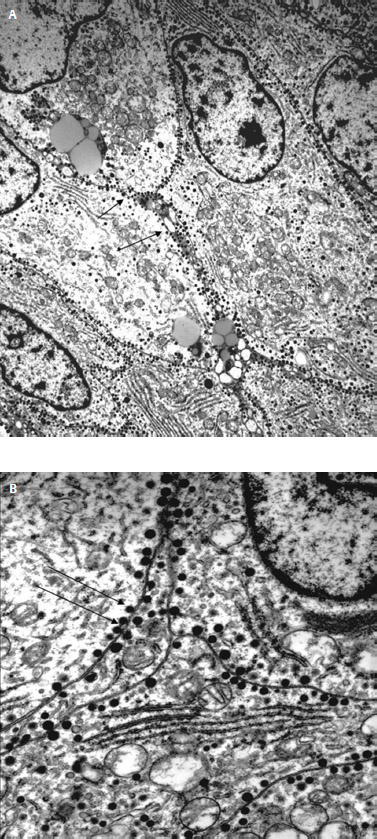
Fig. 12.2 (A) Low-power and (B) high-power electron microscopy demonstrating small, round, evenly dense secretory granules along the cytoplasmic membrane (arrows).
Molecular Pathology
The anterior pituitary transcription factor Pit-1 is required for the growth of somatotrophs, lactotrophs, and thyrotrophs. Mutations in Pit-1 result in the lack of expression of these three cell lineages in both humans and transgenic organisms.10,11 Pit-1 expression and the development of pituitary tumors have been explored in both humans and in the laboratory. Although Pit-1 overexpression can result in the development of GH, PRL, and TSH adenomas, the Pit-1 gene itself may not demonstrate any mutation.12–15
Several oncogenes have been investigated in relation to TSH adenomas but none has consistently been found to be abnormal. The gsp oncogene is expressed in approximately 40% of GH adenomas; however, in Beck-Peccoz et al’s5 review of TSH adenomas that included 11 specimens, none was found to express gsp. The gsp oncogene is responsible for production of G-protein subunits, and thus it has been suggested that disruption of guanosine triphosphatase activity is unlikely to be the underlying pathologic mechanism of TSH adenomas.5
Oncogenes such as c-myc, c-fos, and c-myb have been implicated in pituitary adenomas. The literature on such mutations in TSH adenomas is sparse, but there appears to be no overexpression of these oncogenes in TSH adenomas.16 Ras oncogene and protein kinase C mutations have been found in pituitary adenomas. Nevertheless, data on mutations in p53, retinoblastoma gene, and 11q13 deletions in the context of TSH adenomas are insufficient to draw any reliable conclusion.
 Presentation
Presentation
The presenting signs and symptoms leading to the diagnosis of TSH adenomas usually relate to thyrotoxicosis. Patients may present with a short history, but the duration of signs and symptoms may be greater than 25 years in some cases and can range in severity from mild to the severe. Symptoms related to tumor size or mass effect include visual field abnormalities, cranial nerve palsies, and headache. One large study of 25 patients from the National Institutes of Health found that headaches were present in 39% and visual fields were impaired in 36%.6 Thyrotoxicosis commonly presents with palpitations, arrhythmias, weight loss, tremor, sweating, and nervousness.6,17 Up to 90% of patients may present with goiter.17,18 TSH adenomas may also be associated with multiple endocrine neoplasia type 1 syndrome; however, the pituitary lesions may be found prior to the systemic disease.19 Familial cases of TSH adenoma have not yet been reported in the literature.17
About 30% of cases have associated dysregulation of another pituitary hormone axis.5 With dysregulation of another hormone pathway, the signs and symptoms of other pituitary disorders may also precipitate diagnosis, such as amenorrhea, galactorrhea, loss of libido, and acromegaly. Despite the presence of signs and symptoms related to dysregulation of another pituitary-hormone axis, the associated hormones may not be elevated on serum hormone evaluation. It has been postulated that in cases without elevated levels of associated hormones the mechanism of dysregulation revolves around compression of the pituitary gland or disconnection of the pituitary-hypothalamic axis.17 Patients may also be asymptomatic and are diagnosed incidentally when undergoing neuroimaging of the brain for other reasons.
Unfortunately, because of the rarity of these lesions, many patients present to neurosurgical services after having undergone a lengthy and invasive course over many years prior to diagnosis. In some series the majority of patients have undergone thyroid surgery for excision of goiter. Some receive radioactive iodine or antithyroid drugs. Earlier studies from the 1980s were more likely to include patients who underwent additional treatment prior to diagnosis. Since the advent of commercially available laboratory assays for TSH, from the 1990s to the present these events are becoming less common.6,17
The consequences of misdiagnosis in the setting of a TSH adenoma can be severe. It has been suggested that a similar mechanism involved in Nelson’s syndrome leading to transformation of pituitary cells after adrenalectomy may occur following thyroidectomy in a patient with a TSH adenoma. In addition, it has been postulated that patients with previous thyroid surgery had more aggressive, fibrous, firm, invasive tumors.17,20 It has also been suggested that reactive TSH adenomas may develop as a consequence of primary hypothyroidism.21,22
 Diagnosis
Diagnosis
Biochemical
The diagnosis of a TSH adenoma requires the appropriate history, physical examination, and radiographic findings as well as multiple biochemical markers. A patient with weight loss, tremor, sweating, palpitations, or a goiter in combination with headaches or visual field or eye movement abnormalities should prompt the clinician to consider a TSH adenoma. A routine panel of pituitary hormone tests should be performed in all patients and includes PRL, GH, insulin-like growth factor-1 (IGF-1), luteinizing hormone (LH), follicle-stimulating hormone (FSH), and adrenocorticotropic hormone (ACTH). There is no one serum biochemical marker or test that can be used solely to diagnose TSH adenomas. In addition, the biochemical parameters used to diagnose TSH adenomas have changed over the years as more sensitive assays have become commercially available. This has undoubtedly led to an increase in the rate of diagnosis of these lesions.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


