1 The Science of Aging
 Introduction
Introduction
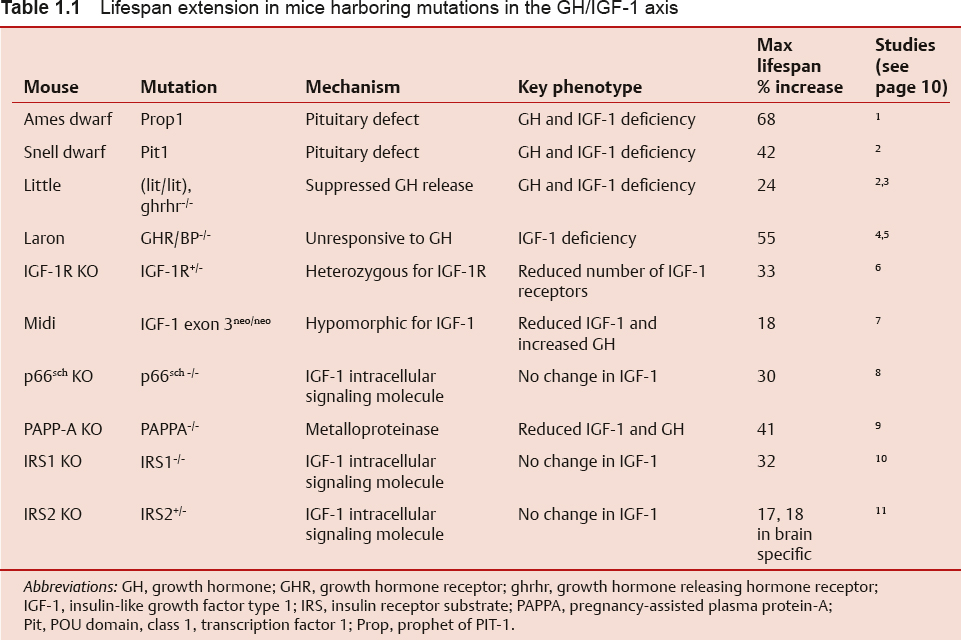
The demographics of the developed world are shifting dramatically due to an increase in life expectancy and a decline in the fertility rate. By the year 2030, the U.S. Census Bureau projects that individuals aged > 65 years will make up almost 20% of the total population.1,2 This represents an increase of 36 million in the > 65 age group, with an increase of 10 million among those aged > 80 years.1 Fig. 1.1 demonstrates the changing survival curve for the U.S. population based on data from the U.S. Social Security Administration projected out to 2100.3 The survival curve becomes more “square” over time, indicating an increased time to first deaths, due largely to a decreased death rate in the first half of life. Mean lifespan increases dramatically, while the increase in maximum lifespan, represented by the final 10% of the population, is far from dramatic. The discrepancy between gains in mean and maximum lifespan can be understood if one considers that early life losses are primarily due to extrinsic factors, the impact of which were diminished over the past century through advances in sanitation and health care and the availability of antibiotics and vaccines.4 The decrease in early life mortality produces the shifts in the survival curves seen in Fig. 1.1. The data for agespecific mortality broken down by 10-year intervals for the U.S. population in 1900, 1939, and 2000 (Fig. 1.2) indicate a reduction in mortality rates at all ages, with the most dramatic reduction in the first 15 years of life.5 Interestingly, for all time periods examined, the lowest mortality rate occurs in the 5 to 14 year age group. The mortality rates increase with increasing age from this time on in all three populations. In contrast to mean lifespan, maximum lifespan exhibits only a modest increase, from 95 to 100 years, between 1900 and 2000. This reflects the fact that the intrinsic aging process has not been impacted to the same degree as the specific pathologies that underlie early life mortality.
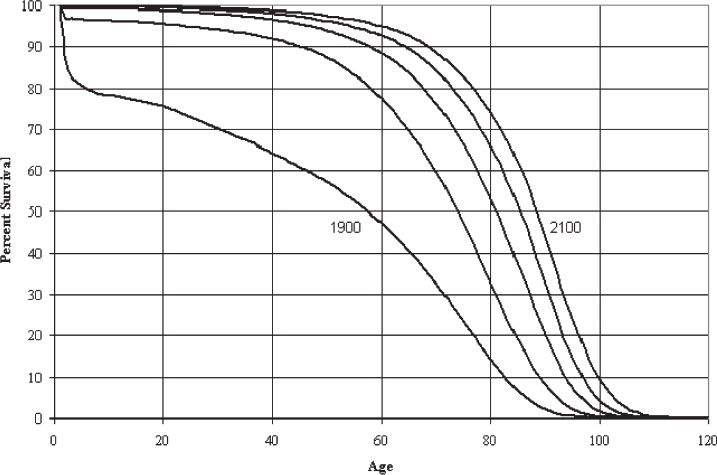
Fig. 1.1 Projected survival curves for the United States in the years 1900, 2000, 2025, 2050, and 2100. (From Bell FC, Miller ML. Life Tables for the United States Social Security Area 1900–2100. Actuarial Study No. 120. Social Security Administration; 2000. http://www.ssa.gov/oact/NOTES/as120/LifeTables_Body.html.)
Interestingly, the effects of the intrinsic aging process can be observed across species. For example, the mortality rate in mice, when examined as a percentage of lifespan completed, is very similar to that in humans (Fig. 1.2), despite a relatively abbreviated mouse lifespan, on the order of 2 to 3 years. Note that the shape of the polynomial trend line for the mouse mortality rate in Fig. 1.3 exhibits a nadir early in life similar to that in humans (~ 10 years in Fig. 1.2). Similarly, a late-life rise in the mortality rate is evident in both species, beginning at ~ 50% of lifespan completed. In humans, this correlates with the 45 to 55 year age range. The similarity in mortality rate curves between the mouse and human suggests that the intrinsic rate of aging is similar in the two species, but it follows a very different trajectory in absolute terms. At the cellular level, the genomic stability of cells from these two species reflects this difference.
Cells from primary explants of tissues from multiple species have been tested for their growth properties.6 Normal cells proliferate for a finite period and eventually undergo a growth arrest known as cellular senescence. The senescence growth arrest is a p53-mediated arrest that appears to serve multiple functions, including tumor suppression and immune activation.7 Interestingly, cells from shorter-lived species such as the mouse demonstrate a greatly reduced genomic stability relative to cells from long-lived species such as humans. Mouse cells escape senescence and generate immortal transformed cell lines that have unstable genomes and exhibit multiple genetic rearrangements.8 In contrast, human cells will transform spontaneously at a rate of less than 1 × 10-6.9 The difference in genomic stability between these species is thought to contribute to differences in the rates of both cancer and mortality.
 Aging Is Characterized by an Increased Vulnerability to Disease
Aging Is Characterized by an Increased Vulnerability to Disease
The most accurate definition of the aging process as it relates to an organism is that aging is characterized by an increasing inability to maintain homeostasis in multiple organ systems, giving rise to an increased vulnerability to disease. Data from the Centers for Disease Control and Prevention support this definition. In Fig. 1.4, deaths per 100,000 population from cancer, heart disease, and cerebrovascular disease are shown as a function of age. Deaths from these diseases increase sharply in middle age and rise progressively thereafter, reflecting the greater vulnerability to multiple diseases in line with an intrinsic aging process. The inability to maintain homeostasis, which contributes to this vulnerability, is evident in the example of age-related changes in lung function. Both forced expiratory volume and response to hypoxia are reduced with age, reflecting a progressive decline in lung function, whereas the loss of reserve capacity in the lungs is an excellent example of the reduced dynamic capacity that characterizes aged organisms.10,11 Underlying the differences in functional parameters are changes in lung architecture due to intrinsic aging. These changes lead to reduced function that requires compensation to maintain normal physiology. Age-related changes and compensatory responses become more pronounced with increasing age, eventually producing the greater vulnerability to disease characterized by the curves in Fig. 1.4. Similar change occurs in all organ systems as a function of age, resulting in the eventual loss of dynamic range and lack of compensatory response typical of the aged organism.
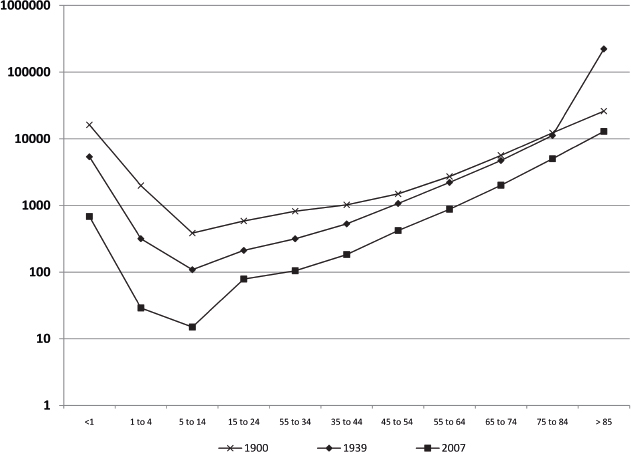
Fig. 1.2 Age-specific mortality rates for the United States in the years 1900, 1937, and 2000. (Calculated from Bell FC, Miller ML. Life Tables for the United States Social Security Area 1900–2100. Actuarial Study No. 120. Social Security Administration; 2000. http://www.ssa.gov/oact/NOTES/as120/LifeTables_Body.html.)
Fig. 1.3 Age-specific mortality rates in a laboratory mouse strain housed under pathogen-free conditions. (Data are recalculated from Lorenzini A, Salmon AB, Lerner C, et al. Mice producing reduced levels of insulin-like growth factor type 1 display an increase in maximum, but not mean, life span. J Gerontol A Biol Sci Med Sci 2014;69(4):410–419.)
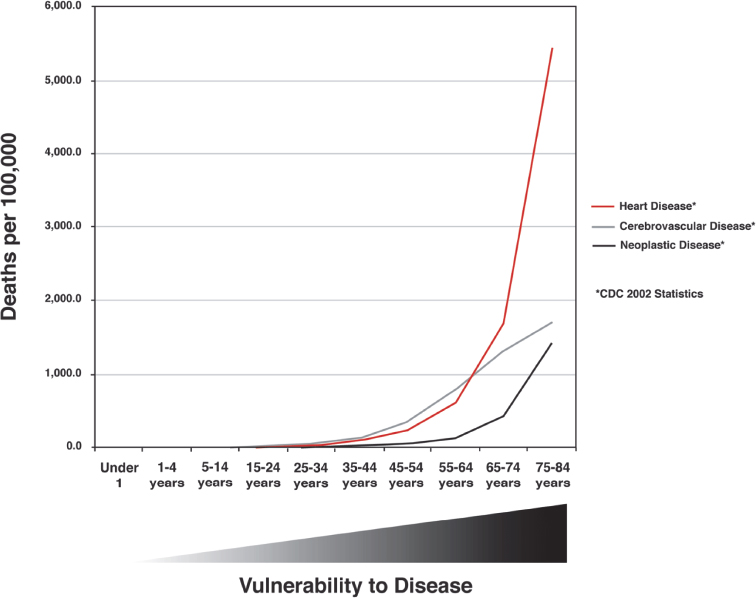
Fig. 1.4 Mortality rates due to all causes, heart disease, cerebrovascular disease, Alzheimer disease, and neoplastic disease in the year 2011 for the United States. Vulnerability to disease is represented beneath the graph. (Data are based on 2011 tables from the Centers for Disease Control and Prevention, Atlanta, GA.)
 Biological versus Chronological Age
Biological versus Chronological Age
Individual variation in the aging process suggests that chronological age may not be the best nor the most clinically relevant measure of the age of an individual. Efforts have therefore been undertaken to identify biomarkers of age that can be used to assess the true age of an individual. Although these efforts have not yet produced any marker or group of markers that can serve this purpose to the satisfaction of the scientific community, the National Institute on Aging continues to support efforts to define biomarkers of aging for their potential clinical impact.12 In addition, systematic approaches to understand individual differences in the rate of aging involve large-scale genetic analyses of populations that enjoy exceptional longevity. The Einstein Ashkenazi Longevity Study in New York has enrolled more than 700 individuals. The New England Centenarian Study at Harvard likewise has also enrolled more than 700 individuals, whereas the Southern Italian Centenarian Study has enrolled more than 900 individuals. Thus far, these groups have identified polymorphisms associated with longevity in the ApoE, IGF receptor, and Lamin A genes.13–15
 Theories of Aging
Theories of Aging
Multiple theories of aging have been proposed. Some suggest that specific types of damage, such as DNA damage or mitochondrial dysfunction, drive the aging process, whereas others examine aging from an evolutionary perspective. The most general question regarding aging asks why organisms age at all. In 1889, August Weismann made the observation that aging is a property of the somatic tissues and not the germline, which possesses an unlimited capacity for self-renewal. The concept Weismann developed stands on two principles: (1) that the germline is immortal, whereas the somatic cells are mortal; and (2) that aging allows for the removal of older individuals to make space for the next generation.16 A synthesis of evolutionary theory and the aging process by Peter Medawar in 1952 led to the concept that aging is not under selective pressure, because reproduction occurs relatively early in life and few organisms in the wild live to advanced age. Thus, aging as a phenomenon is restricted to protected environments and is likely due to an inability to repair damage beyond the reproductive phase of life.17 The concept that aging occurs in postreproductive organisms has led to the theory of antagonistic pleiotropy.18 This theory states that mutations which benefit an organism early in life may have negative effects later in life. According to antagonistic pleiotropy, mutations that increase reproductive fitness may contribute to age-related declines in the same organism. For example, a rapid developmental rate may enhance reproductive fitness in a subset of organisms within a population, but this advantage may lead to increased cancer risk in these individuals as they age.
Building on the concept of a tradeoff between fitness and longevity inherent in the antagonistic pleiotropy theory and based on the idea that an organism must allocate limiting resources to either maintenance or longevity, Thomas Kirkwood developed the disposable soma theory of aging.19 The disposable soma theory postulates that insufficient energy is available to the organism for the maintenance of both germline and soma, forcing an allocation of resources that leads to the eventual deterioration of the somatic tissues. The theory has traditionally centered on the allocation of energy in the form of adenosine triphosphate (ATP); however, we have developed the concept that time is the most likely limiting resource that constrains longevity.20,21 In this iteration of the disposable soma theory, the time allotted for repair of genomic damage is limited due to evolutionary pressure, and it is time, rather than energy, that represents the limiting resource for an organism.
Recently, the possibility that aging may be subject to a level of regulation has been put forth. Trivial arguments have been made based on the fact that single gene mutations can influence longevity, but more rigorous arguments have been posed by population biologists.22 These arguments propose that aging of individuals is an altruistic trait that may lead to enhanced overall fitness of the population.23
Several theories proposing the accumulation of specific types of damage as a primary cause of aging have gained widespread attention. These theories are properly placed under the umbrella of the disposable soma theory because they deal with potential mechanisms whereby the soma may become dysfunctional with age. One of the more prominent of these wear and tear theories is the free radical theory of aging championed by Denham Harman in the 1950s. At this time, the role of the oxygen free radical as a mediator of radiation damage was discovered, and it was postulated that the accumulation of free radical damage might underlie age-related functional decline.24 Because mitochondria are believed to be the primary source of free radicals within the cell, the free radical theory has been intertwined with the mitochondrial theory of aging, which postulates that mitochondrial-generated free radicals are a major source of age-related damage.25 However, free radicals can be generated by other cellular organelles such as peroxisomes,26 and mutations inducing mitochondrial dysfunction have been found to increase lifespan in some cases, generating discussion as to the true role of mitochondrial dysfunction in aging.27 Similarly, DNA damage has been put forth as the primary cause of aging.28 There is evidence for an accumulation of DNA damage with age,29 and mutations that decrease DNA repair capacity induce premature aging, but it has recently become clear that DNA damage responses are more important in aging than DNA damage per se.30 Some arguments have implicated other types of damage, such as glycation end products (caused by nonenzymatic protein linkage of carbohydrates) as primary drivers of aging. But, rather than the absolute level of damage, it is likely that the balance between the accumulation of damage to macromolecules or organelles and the integrity of the complex repair and clearance mechanisms that exist to remove this damage is critical to the aging process.
 Increased Longevity Resulting from Single Gene Mutations
Increased Longevity Resulting from Single Gene Mutations
A remarkable series of experiments has demonstrated that longevity can be influenced strongly by single gene mutations. The insulin-like signaling (ILS) pathway has emerged as a major mediator of longevity in multiple species. In mammals, the ILS pathway is mediated through the binding of insulin-like growth factor type 1 (IGF-1) to the IGF-1 receptor (IGF-1R) in the plasma membrane. Although IGF-1 is expressed in almost all tissues in an autocrine and paracrine fashion, the majority of circulating IGF-1 is released into the circulation by the liver. The regulation of IGF-1 expression is intimately linked to growth hormone (GH) signals emanating from the neuroendocrine system. GH is released into the blood by the pituitary gland in response to growth hormone releasing hormone (GHRH) produced in the hypothalamus. After IGF-1 is produced, it feeds back on the hypothalamus and suppresses GHRH, thereby completing part of a regulatory circuit known as the somatotropic axis. Six IGF-1 binding proteins (IGFBPs) are involved in regulating IGF-1 bioavailability. The major carrier for IGF-1 in the blood is IGFBP3, which combines with a protein known as the acid labile subunit to form the circulating IGF-1 complex. The remaining IGFBPs are expressed in a tissue-specific manner and are important for bioactivity in specific tissues and cells. The specific IGFBPs expressed can affect cell responses to IGF-1, including whether to adopt a proliferative or migratory phenotype, for example.31
IGF-1 is a major determinant of body size during prenatal growth and development. In mice, the IGF-1 knockout and IGF-1/IGF-1R double knockout are lethal to neonates, which show reductions of 60% and 45% in birth weight, respectively, compared with normal pups.32 The influence of IGF-1 on growth in humans is similar. In one case report, a patient homozygous for IGF-1 deletions in exons 4 and 5 was 40% of normal weight at birth, with continued growth retardation at 15 years.33 In Laron syndrome, a type of dwarfism, the GH receptor is mutated and unable to stimulate IGF-1 expression. Children born with this disorder exhibit reduced GH signaling and present with several growth-related defects, including small organ size, weakened hair and nail growth, and sarcopenia.34 There are no reports indicating that patients with Laron syndrome experience increased lifespan; however, mice modeled after the Laron mutation, through ablation of the GH receptor, are dwarfs and exhibit a significant increase in lifespan (Table 1.1).
The deleterious effect that a severe loss of IGF-1 or its receptor confers on viability in mice is in remarkable contrast to low to moderate reductions in ILS. Kenyon et al first reported that a hypomorphic mutation in the Caenorhabditis elegans IR homologue Daf-2, increases lifespan. The worms harboring this mutation achieved a 300% increase in lifespan.35 Studies in Drosophila expressing a hypomorphic IR showed an 85% increase in lifespan as well as decreased body size.36 The IR homologues in worms and flies both respond to a variety of “insulin-like peptides” and are evolutionarily diverged from the IR and IGF-1R in vertebrates, which respond to distinct hormone ligands. Despite these differences, ILS is a highly conserved intracellular signaling pathway and transgenic mouse models that express reduced levels of IGF-1 or that are heterozygous for the IGF-1R achieve a range of small to modest increases in lifespan accompanied by dwarf phenotypes (Table 1.1). Mouse models such as the Ames dwarf that achieve a reduction in circulating IGF-1 through impaired GH production reach the longest lifespan, sparking debate as to whether GH signaling has an independent role in lifespan extension that enhances reduced ILS. Fibroblasts derived from the long-lived Ames and Snell dwarf mice, for example, exhibit resistance to H2O2, paraquat, and ultraviolet (UV) light.37 Cells from other long-lived mouse models have also been assessed and exhibit stress resistance to various toxins and oxidants as well.38 Thus resistance to stress appears to be an important factor contributing to the increased lifespans in these mice.