pupillae. Cholinergic discharges from their muscarinic end plates result in miosis and accommodation. The dilator muscle is innervated by third-order, postganglionic sympathetics originating from the superior cervical ganglion. These nerve fibers surround the internal carotid artery as a plexus in the cavernous sinus from where they emerge to enter the orbit with the nasociliary branch of the ophthalmic nerve, a division of the trigeminal nerve. The nasociliary nerve gives off several long ciliary nerve branches that bypass the ciliary ganglion and travel with the short ciliary nerves before supplying the dilator pupillae. Their adrenergic discharges result in mydriasis. Both autonomic groups pass through the ciliary ganglion, a small body of variable size and shape that lies within orbital fatty tissue, posterolateral to the optic nerve and medial to the lateral rectus muscle. Only the cell bodies of postganglionic parasympathetics lie within this ganglion; vasomotor and sensory sympathetic fibers from the nasociliary nerve traverse the ganglion to emerge in the short posterior ciliary nerves without synapsing.
to regulate the amount of light reaching the retina;
to diminish the chromatic and spherical aberrations produced by the peripheral imperfections of the optical system of the cornea and lens; and
to increase the depth of field (analogous to the f-stop setting of a camera).
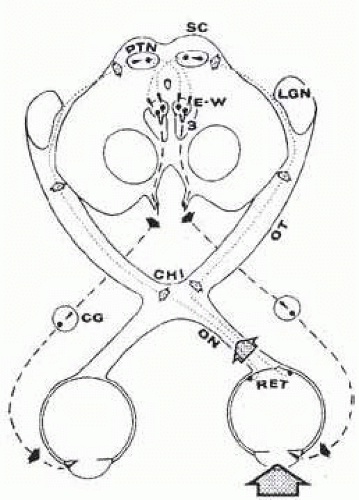 FIG. 15.1 Pupillary light reflex. Light in left eye (dotted arrow) stimulates retina (RET), whose afferent axons (fine dashed lines) ascend optic nerve (ON), decussate at chiasm (CHI), and terminate in pretectal nuclear complex (PTN). Lateral geniculate nucleus (LGN) is bypassed by these pupillomotor fibers. The PTN is connected by crossed and uncrossed intercalated neurons to both Edinger-Westphal parasympathetic motor nuclei (E-W), which comprise the dorsal aspect of the oculomotor nuclear complex.3 Preganglionic parasympathetic fibers (heavy dashed lines) leave ventral aspect of midbrain in the substance of the third cranial nerves. After synapsing in the ciliary ganglia (CG), the postganglionic fibers innervate the pupillary sphincter muscles. Note that uniocular light stimulus evokes bilateral and symmetric pupillary constriction. Brain stem diagram represents section through level rostral to superior colliculi (SC). |
scatter and focusing defects have an increasing effect with larger pupils. In addition to autonomic control, the size of the pupils is influenced by the physical integrity of the iris, intensity of retinal illumination, the near-effort reflex, the state of retinal light adaptation, and supranuclear influences from the frontal and occipital cortex above the pretectal area and from the reticular formation of the brain stem below. At a given moment, any or all of the aforementioned factors variably influence pupillary size and reactivity. Thus in the awake state, the pupil is in constant motion, a condition of physiologic unrest called hippus. Although it is described in diverse conditions ranging from encephalitis to schizophrenia and from cataracts to hemorrhoids, this incessant change in pupil size is of no pathologic significance.5
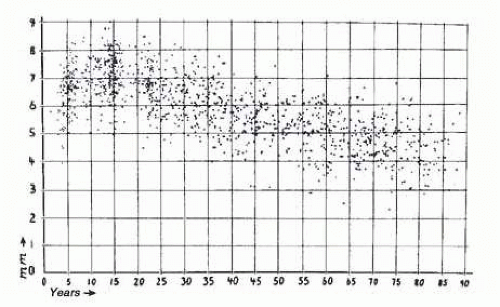 FIG. 15.2 Pupillary size in darkness of 1,263 subjects chosen at random; average pupil size was used ([R+ L]/2). Abscissa shows horizontal diameters in millimeters, ordinate shows subjects’ age in years. Note the wide scatter but obvious age trend. See also Figure 15.3 (top curve). (Reprinted with permission from Loewenfeld IE. Pupillary changes related to age. In: Thompson HS, Daroff RB, Frisen L, et al, eds. Topics in Neuro-ophthalmology. Baltimore, MD: Williams & Wilkins; 1979:129.) |
which newer devices are compared. It affords more accurate measurements and documentation of the pupil diameter and reactivity. Numerous pupillometry devices are commercially available for specific uses and are less cumbersome than the original research tools. They have variable sources of illumination, spatial and temporal resolution, can be monocular or binocular, and have additional features such as detection of sleep waves and eye movements (Fig. 15.4). A practical clinically useful pupillometer will need to satisfy requirements such as portability, simplicity of use, reproducibility of measurements, technician-independence or automaticity, variable illuminations, and the capacity to reliably negate influences from accommodation, convergence, and consensual response. A study comparing the readily available techniques found that digital photography and the handheld Colvard monocular pupillometer (Oasis Medical Inc.) compared most favorably to infrared pupillographic pupil size measurements when compared with rulers and templates.10
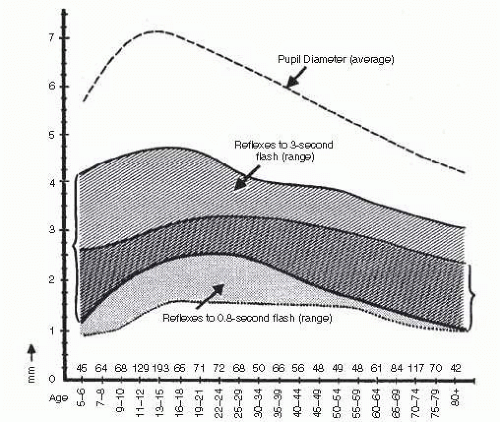 FIG. 15.3 Normal ranges of light reflex amplitude for long and for short flashes. Shaded area (left bracket) is normal range for 3-second flashes; stippled area is normal range for 0.8-second flashes. The numbers above the abscissa indicate the number of subjects per age group. Note the early peak, followed by decline with age for reactions to long light flashes. In contrast, reflexes elicited by short flashes show relatively flat age curve. (Reprinted with permission from Loewenfeld IE. Pupillary changes related to age. In: Thompson HS, Daroff RB, Frisen L, et al, eds. Topics in Neuro-ophthalmology. Baltimore, MD: Williams & Wilkins; 1979:137.) |
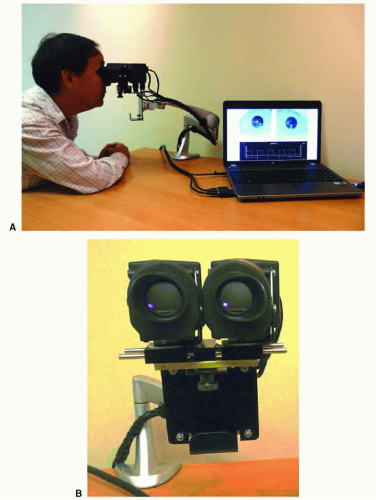 FIG. 15.4 Pupillometer (A) with close-up front view (B). In the past, most computerized pupillometers were designed primarily for clinical research. More patient-friendly pupil testing devices are now becoming available. Such instruments would optimally be compact, binocular (allowing both direct and consensual pupil responses to be recorded for each light stimulus), have a built-in light stimulus and should be easy to use in testing, with little setup time. Chromatic light stimuli (e.g., red and blue) might be advantageous to use in order to differentiate photoreceptor disorders from inner retina and optic nerve disorders. These devices are more suitable for routine clinical use to assess afferent input from the retina and optic nerve as well as efferent output abnormalities, manifesting as anisocoria. These pupillometers might be used as a screening test for a RAPD and anisocoria. The instrument shown above uses a compact optical “head” on an articulating arm that consists of two miniature video cameras and a bank of light-emitting diodes (LEDs) for each eye for light stimuli. The LEDs consist of an array of red, green, blue, and white lights that can be independently turned on over a 5-log unit range of intensities. A diffuser is used so that each light stimulus covers at least 20° of visual field and with scatter of light, even a greater area at brighter light intensities. The instrument can be run by a portable or desktop computer with software that allows one to custom-configure different clinical tests or use those provided by the company (Neuroptics, Inc.). Software will automatically analyze recorded pupil movements and will provide a report. (Courtesy Dr. Randy Kardon.) |
TABLE 15-1 Outline of Physiologic Reflexes of the Pupil: the Stimuli, Effects, and Proposed Autonomic Mediation | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
(from nasal retinal receptors of the contralateral eye) and uncrossed fibers (from temporal retinal receptors of the ipsilateral eye). In the posterior aspect of the optic tract (pregeniculate), the retinomesencephalic pupillomotor branches of the afferent axons gain the pretectal nuclear area by transversing the brachium of the superior colliculus into the rostral midbrain. Intercalated neurons interconnect from the preolivary nucleus of the posterior commissure and lentiform nucleus21 to the EWN by crossing dorsal to the aqueduct in the posterior commissure and by coursing ventrally in the periaqueductal gray matter. This simplistic anatomic approach belies the true complexity of the neuroanatomy and neurophysiology of the pretectal nuclear complex. The reader is referred to articles by Smith et al.,21 Breen et al.,22 Carpenter and Pierson,23 Benevento et al.,24 Burde,25 and Loewenfeld’s1 textbook.
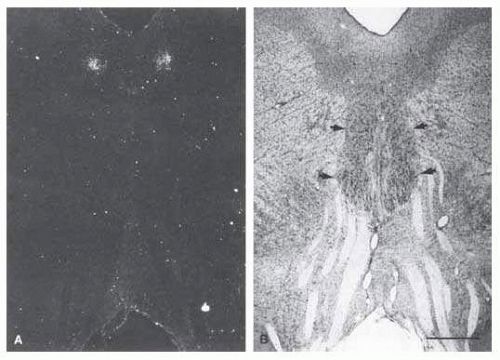 FIG. 15.5 A: Transneuronal autoradiographic label in the Edinger-Westphal nuclei seems bilaterally adjacent to the midline, ventral to the cerebral aqueduct. B: The label seen in (A) corresponds on each side to the fairly distinct cell group (thin arrows), the lateral visceral cell column of the Edinger-Westphal nucleus, shown in a Nissl-counterstained section. The somatic subnuclei of the oculomotor complex (thick arrows) contain larger nuclei. Fascicles from the oculomotor complex are seen streaming inferiorly toward the interpeduncular fossa. Scale bar = 1 mm. (Reprinted with permission from Kourouvan HD, Horton JC. Transneuronal retinal input to the primate Edinger-Westphal nucleus. J Comp Neurol. 1997;381:68.) |
Using multiple markers, including wheat germ agglutinin, May and Fratkin30 have further isolated the EWN in the macaque monkey and deduced that it is composed of a solitary column of cells. Increasingly sophisticated studies such as functional magnetic resonance imaging (MRI), positron emission tomography, and in situ hybridization should help to elucidate the detailed architecture and physiology of this nucleus.
Excitatory: retinomesencephalic (light stimulus) and occipitomesencephalic (near reflex) and
Inhibitory: corticomesencephalic and hypothalamomesencephalic pathways and the ascending reticulomesencephalic system.
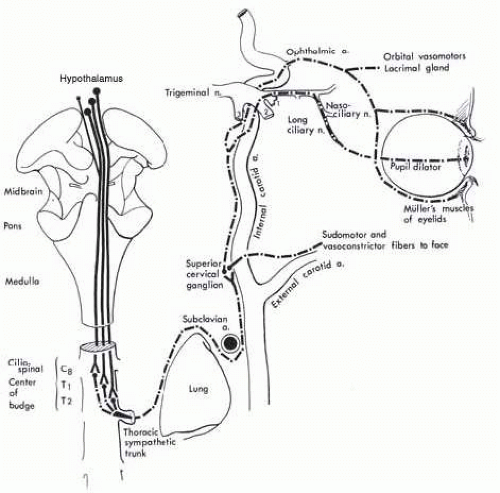 FIG. 15.6 Ocular sympathetic pathways. Hypothalamic sympathetic fibers comprise a polysynaptic system as they descend to the ciliospinal center. This intra-axial tract is functionally considered the “first-order neuron.” The second-order neuron takes a circuitous course through the posterosuperior aspect of the chest and ascends in the neck in relationship to the carotid system. Third-order neurons originate in the superior cervical ganglion and are distributed to the face with branches of the external carotid artery and to the orbit via the ophthalmic artery and ophthalmic division1 of the trigeminal nerve. |
fibers to the tympanic plexus of the middle ear and petrous bone,
fibers temporarily joining the path of the intracavernous abducens nerve before anastomosing with the first division of the trigeminal nerve,
anastomoses with the ophthalmic-trigeminal nerve (the primary pupillomotor pathway via the nasociliary nerve), and
fibers surrounding the ophthalmic artery and ocular motor nerves at the level of the cavernous sinus.
orbital vasomotor,
pupillary dilators,
smooth muscles of the upper and lower lids (Müller muscle),
the lacrimal gland, and
trophic fibers to uveal melanophores.
of regard, a “near synkinesis” is evoked that includes (1) increased accommodation of the lens, (2) convergence of the visual axes of the eyes, and (3) pupillary constriction. Many theories have been proposed to explain how accommodation enables near vision but von Helmholtz theory remains the most widely accepted.40 He proposed that contraction of the ciliary muscles cause the zonules to relax resulting in a more globular convex lens and anterior rotation of the lens-iris diaphragm. This change in architecture alters the focal point of the lens, allowing the eye to focus clearly on near objects.
When there is a significant unilateral or asymmetric afferent visual pathway disruption caused by optic nerve or widespread retinal disease, the pupils show a subnormal response to light stimulation of the eye with the greater field or (generally) acuity loss. The pupils have a more extensive constriction response with light stimulation of the normal or less involved eye. It is this combination of subnormal direct pupillary light response and a normal indirect (consensual) response when the opposite eye is illuminated that constitutes the RAPD.
TABLE 15-2 Characteristics of Pupils Encountered in Neuroophthalmology | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
or penlight. During the test, the patient must look at a distant fixation target to avoid accommodative miosis.
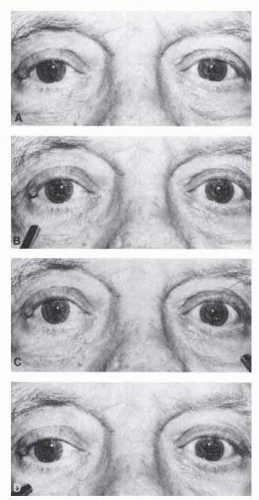 FIG. 15.7 Swinging flashlight test for afferent pupil defect. The patient is a 72-year-old man with right visual loss due to ischemic optic neuropathy. A: Pupils are equal in dim light. B: Illumination of right eye results in modest bilateral constriction. C: When the light swings to the left, there is more extensive constriction in both pupils. D: When the light swings back to the right, both pupils dilate. |
has occurred or as a prognostic indicator for final visual recovery. In a small series, Alford et al.56 found that patients with traumatic optic neuropathy and an initial RAPD of log 2.1 or greater showed minimal visual recovery even after megadose intravenous steroid therapy. Those with RAPD measurements less then log 2.1 recovered to visions of 20/30 or better. Younis and Eggenberger examined 72 patients with unilateral or asymmetric demyelinating optic neuropathy and found significant linear correlation between retinal nerve fiber thickness, measured with ocular coherence tomography (OCT) and RAPD, measured in log units. These findings indicate that the log unit value of RAPD may be used to predict the extent of retinal nerve fiber layer (RNFL) loss in this type of neuropathy.57
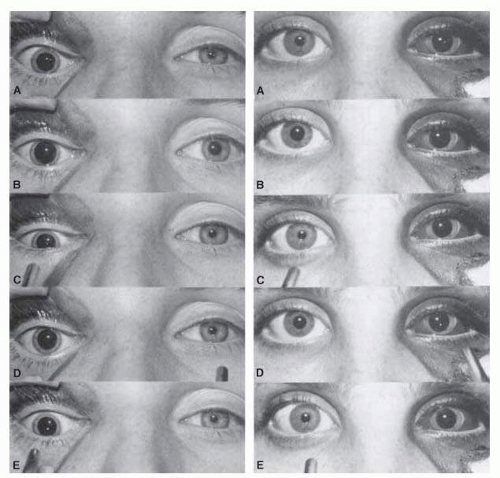 FIG. 15.8 Swinging flashlight test in two patients with mydriasis on side of orbital trauma. Pupils in bright (A) and dim (B) room lighting reveal normal responses on the uninjured side and minimal, if any, response on the affected side. As the flashlight is swung from right (C) to left (D) and again to right (E), one can observe the normally reactive pupil. The patient on the left has no afferent pupillary defect, whereas the patient on the right has a left RAPD. |
reflect optic neuropathy due to demyelination, ischemia, compression, or asymmetric glaucoma.
necessarily all of the time. Using photographic techniques, Lam et al.79 determined pupil size in 128 healthy individuals, measuring greatest pupil diameter twice each day for 5 consecutive days. Forty-one percent showed anisocoria of 0.4 mm or greater at one time or another and 80% showed anisocoria of 0.2 mm or greater at some time. A majority of the subjects, therefore, had anisocoria of some degree at one time or another (Fig. 15.9). Anisocoria is as common in men as in women, in the morning as in the afternoon, in dark as in light irides, and in the young as in the aged (although Loewenfeld80 suggested that anisocoria is more common in the elderly). In a study of healthy neonates, anisocoria of 0.5 mm or more was found in about 20% of infants, but none had anisocoria greater than 1 mm.81 The physician can detect regularly a pupil difference of as little as 0.2 mm in diameter, given that clinical judgment of inequality is based more on impressions of pupil area than on measured diameter. Because the percentage difference in area is greater for smaller than for larger pupils, for any given diameter difference, anisocoria is detected more easily in smaller pupil pairs. The prevalence of anisocoria decreases in bright conditions when measured as a difference in pupil diameter but not when it is assessed as a ratio of pupil areas.82
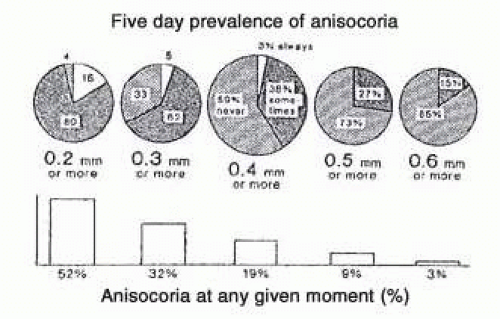 FIG. 15.9 Prevalence of simple anisocoria. (From Thompson HS. The pupil. In: Lessell S, van Dalen JTW, eds. Current Neuroophthalmology. Chicago, IL: Year Book Medical Publishers; 1989:214.) |
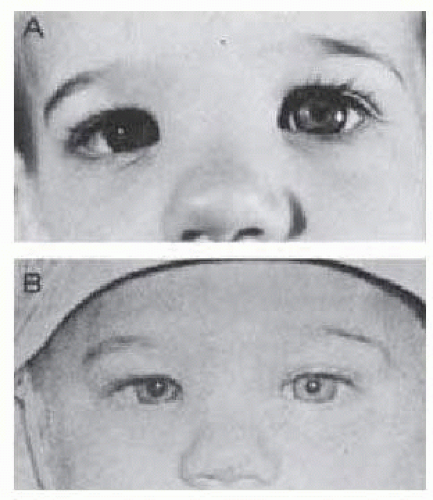 FIG. 15.10 A: Anisocoria noted at age 14 months in otherwise healthy infant. B: Photograph at age 6 months confirms chronicity and benign nature of finding. There was no iris heterochromia. |
affect the pupil size disparity. Generally, if anisocoria is greatest in the dark, the abnormal pupil is the miotic one. Conversely, if anisocoria is greatest in the light, the abnormal pupil is the mydriatic one. Because the etiology of anisocoria may range from abnormal iris structure to disruption of pupillomotor pathway and supranuclear integrity, it is imperative to take a comprehensive history and evaluate the patient for all levels of disruption.
TABLE 15-3 Causes of Anisocoria and Changes with Illumination | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
TABLE 15-4 Clinical Approach to the Patient with Anisocoria | |||||
|---|---|---|---|---|---|
| |||||
been reported in association with concomitant stationary night blindness and congenital achromatopsia and subsequently with other retinopathies and optic neuropathies.86
the tonic pupil. With the passage of time, the anisocoria becomes less marked as the initially larger tonic pupil gradually becomes less dilated and eventually even miotic over the years93, 99 (Fig. 15.11).
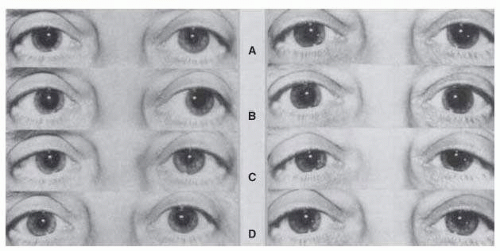 FIG. 15.11 A: A 52-year-old woman with right tonic pupil (left); 1 year later, involvement of the left pupil developed as well (right). Pupils are shown in bright (A) and dim (B) room lighting after near-convergence attempt (C), and in dim room lighting after instillation of 0.125% pilocarpine in both eyes (D). Note that the right pupil is smaller 1 year later (A) (right) than when it was “fresh” (A) (left). With time, the right pupil has also become more responsive to near-accommodation effort (C) (left and right). Parasympathetic hypersensitivity is seen in the right pupil (D) (left) and 1 year later bilaterally (D) (right). |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


