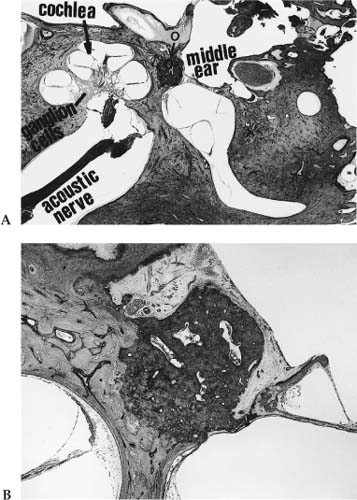
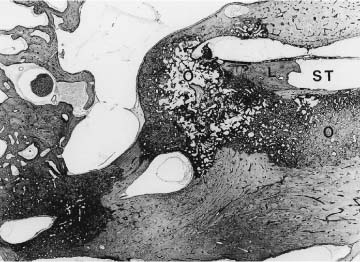
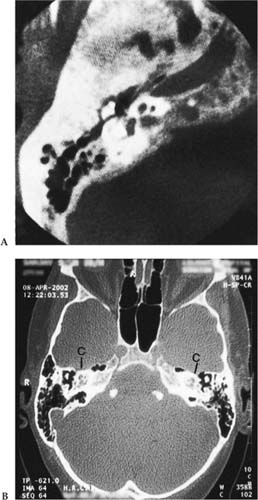
Chapter 2 Otosclerosis is a primary and exclusive disease affecting only the otic capsule and the ossicles. It is a localized disorder of bone metabolism of the otic capsule avascular endochondral bone that is characterized by disordered resorption and deposition of bone. Otosclerosis occurs only in the temporal bone (Wang et al 1999). The exact etiology of otosclerosis is not known at this time, although many theories have been postulated. None of these theories have been proven to be a definite cause of otosclerosis. Postulated etiologies are hereditary factors, endocrine and metabolic factors, and vascular factors. Alterations in vascularity (either an increase or a decrease in blood supply) have been postulated by Witmaack (1930), Wolff (1950), and Mendoza and Ruis (1966) to play a role in the etiology of otosclerosis. Intrinsic and extrinsic mechanical stresses as the result of an erect posture were once considered (Mayer 1917) as a cause of otosclerosis, because the petrous pyramid is located at the base of the skull. Because it was located at the base of the skull, it was subjected to mechanical strains exceeding the physiological limits of tolerance, which resulted in defects in the endochondral layer of the bony labyrinth. It was further theorized that the stress factors appeared during the phylogenic and otogenic development due to rotation of the petrous bones at the base of the skull. It was thought that the cochlea moved forward and upward and the vertical semicircular canals backward and downward. Thus, the petrous pyramid has an almost horizontal position in humans versus a vertical position in quadrupeds. This theory was abandoned because otosclerotic changes first appeared in the footplate of the stapes. Fowler (1949) suggested that otosclerosis might be an expression of a general mesenchymal hypoplasia. This was further supported by Ogilvie and Hall (1962), who postulated that otosclerosis was a local manifestation of osteogenesis imperfecta. In recent times viral infections and autoimmune factors have also been postulated to trigger the otosclerotic processes. Ruedi (1963) demonstrated shunts between the vascular system of the otosclerotic bone and the inner ear, and suggested that venous stasis from these shunts might be responsible for sensorineural hearing loss. It is difficult to assess the true onset of otosclerosis because the development of histopathologic changes is gradual. Clinical otosclerosis is commonly seen between the ages of 30 and 40 (Nager 1969), with the average age of presentation being 33. DeJuan (1960), comparing the age of onset among various age groups, noted that 28% of cases occurred between ages 18 and 21, 40% presented between ages 21 and 30, and 22% presented between ages 31 and 40. The clinical presentation of otosclerosis is usually that of a conductive hearing loss, which may be unilateral or bilateral. Occasionally it may present as a sensorineural hearing loss. The onset of hearing loss is usually between the fourth and fifth decades, with a higher prevalence in women than men (in the ratio of 2:1). Hearing impairment reaches its maximum in the third decade and then usually remains stable (Glorig and Gallo 1962). The prevalence of otosclerosis has been reported as 0.1 to 1.0%, with an average of 0.3% (Gordon, 1989). Autopsy studies (Konigsmark and Gorlin 1976) have found histologic otosclerosis in 5 to 18% of the general population. Jahn and Vernick (1986) stated that 10% of Caucasians develop histo-logic otosclerosis, and 1% go on to develop clinical otosclerosis. Shambaugh (1961) found that histological otosclerosis was 10 times more common than clinical otosclerosis. Friedmann (1974) and Morrison (1967), however, found the incidence to be approximately 2%. It can therefore be seen that the exact incidence of clinical otosclerosis is not clear and next to impossible to determine. Many authors have noted that in recent years the incidence of patients presenting with otosclerosis has fallen dramatically. The prevalence varies in racial populations, being rare, for example, in African blacks and Asians. There is a definite racial predisposition. Otosclerosis is more commonly found in Caucasians. Clinical otosclerosis has been reported to be present in 1% of the population of the United Kingdom, occurring mostly among white females (Hinchcliffe 1961). Guild (1944) reported histologic evidence of otosclerosis in 18.5% of middle-aged white women, 9.7% of adult white men, and only 1% of adult blacks. Cawthorne (1952) found that otosclerosis was more prevalent in fair-haired men than in dark-haired men. The prevalence of otosclerosis is low among Asians (Altmann et al 1967; Nakamura 1968). The prevalence of otosclerosis is low among Japanese (Goto and Omori 1957; Horiguchi 1953; Takahara et al 1959), Chinese, and Indonesians (Nizar 1960). In a comparative study conducted in Hawaii, Joseph and Frazer (1964) found the incidence of otosclerosis to be more prevalent in Caucasians than in Japanese. Rosen and coworkers (1962) did not find clinical cases of otosclerosis in Sudan. Among the Todas in India, however, the prevalence of otosclerosis was estimated at 17%. Kapur and Patt (1966) emphasized the presence of consanguineous marriages, a custom among the Todas. Consanguinity would then appear to influence the data retrieved. The prevalence of otosclerosis in Native Americans is extremely low across the North American continent (Cambon et al, 1965). Wiet (1979) evaluated a large number of Native Americans and found that only 10 had undergone stapedectomy, of which only three had confirmed otosclerosis, a number representing a minute fraction of the entire population examined. Tato and Tato (1967, 1969) evaluated 5000 Native Americans and found no cases of otosclerosis at all. When there is a racial mixture, however, otosclerosis begins to manifest itself (Goycoolea 1991). Otosclerosis is thought to be more prevalent in women. Schmidt (1933) quoted a female preponderance of 72.5% incidence, whereas Shambaugh (1952) found an incidence of 68% and Cawthorne (1955) an incidence of 67% of women who developed otosclerosis. Because otosclerosis is not a genetically sex-linked characteristic, a ratio of 1:1 would have been expected. However, this has not been found to be true. Endocrinologic factors have been suspected in women. Hueb et al (1991) found a higher incidence of bilateral otosclerosis in women than in men, thus prompting them to believe that women would be more likely to seek medical advice than men. This might possibly account for the apparent gender predisposition. Otosclerosis becomes evident during the childbearing years. Most authors report otosclerosis becoming aggravated during pregnancy. Shambaugh (1967) analyzed 475 female patients and reported that 50% did not find any hearing impairment following pregnancies, whereas 8% noticed hearing impairment immediately following pregnancy. In the remaining 42% of patients, hearing loss was associated with pregnancy. Thus, in Shambaugh’s series there is a 50% chance of an association of hearing loss following pregnancy. Repeated pregnancies in the same woman, however, did not further worsen hearing. Walsh (1954) found no evidence of a relationship between hearing loss, pregnancy, and otosclerosis. Shambaugh (1967) estimated that the risk of hearing loss to a woman following pregnancy is approximately 1 in 24. Elbrond and Jensen (1979) and Gristwood and Venables (1983) also noted the association of hearing loss following pregnancy. Elbrond and Jensen (1979) studied the hearing threshold of 144 women before and after stapedectomy. The women were between 16 and 40 years old, and all had undergone stapedectomy for otosclerosis. The observation period for the women who became pregnant following stapedectomy was between 4 years and 9 months and 5 years and 8 months. The authors observed that (1) hearing loss became greater in those who became pregnant than in those who did not and (2) the hearing loss became significantly greater in the nonoperated ear of those patients who became pregnant following surgery. It would thus seem that stapedectomy confers some protection from further hearing loss following pregnancy. Although these authors note a correlation between pregnancy and the onset of hearing loss following pregnancy, they are not clear on how this occurs. Morrison (1979) attributed this to increased estrogen levels, which cause fragility of the lysosomal membranes, with the consequent release of enzymes that in turn activate the otosclerotic process. Causse and Causse (1991) postulated that the otosclerotic process is stimulated by the release of increased quantities of estrogen. The increase in estrogen levels causes the lysosomal membranes to become fragile, which in turn leads to the rupture of the membranes with the consequent diffusion of their enzymes, resulting in the destruction of the cochlea and its structures. A progressive conductive hearing loss in adults is typical of the way otosclerosis presents (Chole and McKenna 2001). This is due to the fixation of the stapedial footplate along its anterior anulus. On rare occasions, otosclerosis can be associated with sensorineural hearing loss (Schuknecht and Kirchner 1974). This is due to cochlear otosclerosis. It is thought that some individuals may present with cochlear otosclerosis in the absence of a conductive hearing loss. Temporal bone studies have shown that involvement of the endosteal portion of the cochlea is associated with sensorineural hearing loss and diminished bone conduction hearing thresholds (Ghorayeb and Linthicum, 1978; Hueb et al, 1991). Hyalinization of the spiral ligament adjacent to the otosclerotic foci has been shown to be associated with sensorineural hearing loss (Antoli-Candela et al 1977; Gussen 1975; Hinojosa and Marion 1987). Although less than 1% of the population develops clinical otosclerosis, the finding of an otosclerotic focus upon autopsy is much more common. Histo-logic otosclerosis (Guild 1944) is a disease process without clinical symptoms or manifestations and can only be discovered by routine sectioning of temporal bones (Fig. 2–1) at autopsy. Histologic otosclerosis may be seen as an incidental postmortem finding without causing clinical symptoms. Clinical otosclerosis is otosclerosis at a site where it causes a hearing loss, which may be conductive, sensorineural, or mixed. Histologic otosclerosis has been reported as 8.3% and 11% in large random autopsy series. Otosclerotic foci can sometimes be seen on high-resolution computed tomography (CT) scans of the temporal bone as areas of hypodensity of the otic capsule or in the vicinity of the oval window (Valvassori 1993). CT scanning, however, is not reliable in detecting otosclerosis, especially when the lesions are sclerotic (Thiers et al 1999). It is possible that future advances in CT imaging and densitometry may be more sensitive in the detection of these lesions. Otosclerotic lesions may also show contrast enhancement on magnetic resonance imaging (Ziyeh et al 1997). Otosclerosis, or otosclerotic-like lesions of the footplate and otic capsule, has been observed in other inherited bone disorders. Otosclerosis is also seen in some patients with osteogenesis imperfecta who commonly develop conductive hearing loss. The lesions in the temporal bone appear similar to those of otosclerosis (Nager 1988), although some patients have lesions consisting of diminished calcification and microfractures. The bony lesions of Paget’s disease in the otic capsule may appear similar to otosclerosis, although their distribution within the temporal bone is distinct (Fig. 2–2). Large multinucleate osteoclasts are much more prominent in Paget’s disease than in otosclerosis. It must be stated that the conductive hearing losses sometimes associated with Paget’s disease are not due to ossicular lesions, but may be due to the loss of mineral density in the otic capsule (Khetarpal and Schuknecht 1990). Paget’s disease of the bone, like otosclerosis, is a disorder of localized bone remodeling, with osteoclastic resorption followed by compensatory increases in bone formation. Paget’s disease affects localized regions of the skeleton while sparing others; however, unlike otosclerosis, it is not restricted to the temporal bone. Thirty percent of those suffering from Paget’s have a family history of the disease, causing speculation about a genetic basis of this disease. There is recent evidence from linkage studies that chromosome 18 carries a predisposition gene for Paget’s disease (Leach et al 1999). In addition, there is compelling evidence of a viral etiology in Paget’s disease (Mills et al 1994). Paget’s disease may cause conductive and/or sensorineural hearing loss by involvement of the otic capsule and ossicles. Otosclerotic-like changes have also been described in CamuratiEngelmann disease (CED) (Chole and McKenna 2001). CED, also known as progressive diaphyseal dysplasia, is a rare disorder of rapid bone turnover, diaphyseal hyperostosis, and muscle hypoplasia. Affected individuals commonly experience mixed hearing loss and vertigo. Radiologically, lesions similar to otic capsule otosclerosis have been seen (Hanson and Parnes 1995; Huygen et al 1996). Figure 2–1 (A) Photomicrograph of a human temporal bone with histologic otosclerosis (O). This focus lies anterior to the oval window and has not resulted in any symptoms. This is an incidental finding on autopsy, discovered accidentally, and is termed histologic otosclerosis. (B) Magnified view of the same human temporal bone section demonstrating histologic otosclerosis. Figure 2–2 Computed tomography (CT) scan (A) of a patient suffering from Paget’s disease. Compare this with a CT scan (B) of a patient suffering from cochlear otosclerosis. The cochlea in (A) is normal, but the cochlea (C) in (B) is lucent. Otosclerotic foci are commonly found in front of the oval window. This location has been reported in 80 to 90% of temporal bones with otosclerosis (Guild 1944). The second most common site is the round window (Fig. 2–3). Very rarely will the round window be occluded by otosclerosis, with complete obliteration of the round window accounting for 6% of temporal bones examined (Hueb et al 1991). Reports of round window involvement range from 30 to 50% (Guild 1944; Nylen 1949). It was also noted, however, that despite being very close to the round window, otosclerotic focus merged in approximately 12% of cases (Nager 1969). The surgical findings of extensive otosclerosis obliterating the round window have been reported to be approximately 1%. Shea and Farrior (1987) reported this finding in 30,000 patients who had undergone stapedectomy. Schuknecht and Barber (1985) reported involvement of the round window in 30% of cases of clinical oto-sclerosis and 17% of temporal bones of histologic otosclerosis. Other sites that were involved were the apical medial wall of the cochlea, posterior to the oval window, the posterior internal auditory canal, the cochlea aqueduct, and the semicircular canals. In the same report, involvement anterior to the oval window as the only focus was seen in 51% of cases of clinical oto-sclerosis. Involvement of the malleus and incus, though uncommon, can cause fixation of the head of the malleus in the attic. Otosclerosis usually affects both temporal bones symmetrically. Nylen (1949) found that 70 to 80% of cases were involved bilaterally and were symmetrical in the areas of involvement. Nagen (1947) found histologically unilateral otosclerosis in 10% of temporal bones. Figure 2–3 Otosclerotic focus (O) at the round window. New bone formation (L) has replaced the round window. ST = scala tympani. Cartilage persists throughout life in certain parts of the otic capsule. Bast and Anson (1949) described seven regions where cartilage has been noted: (1) fissula ante fenestram, (2) fossula post fenestram, (3) intracochlear area (endochondral layer), (4) cochlear area (round window), (5) semicircular canals, (6) petrosquamous suture, and (7) base of the styloid process. The otic capsule consists of three layers: the endochondral, the periosteal, and the endosteal. The endochondral layer is located lateral to the endosteal layer and medial to the periosteal layer. It contains areas of calcified cartilaginous matrix, and occasionally cartilage cells remain. The calcified areas have capillary buds. Osteoblasts deposit bone in the lacunae, forming small bony globules, or globuli ossei. The primitive perichondrium (Bast and Anson 1949) becomes the periosteum or the periosteal layer. The fissula ante fenestram is located anterior to the oval window and is the most common site for otosclerosis. In 70 to 90% of cases, otosclerotic lesions may replace the fissula ante fenestram (Nager 1969). The fissula ante fenestram forms a fibrous connection between the periotic tissues and the tissues of the middle ear. The fissula reduces in size when a new secondary cartilage is produced from the perichondrium. The fossula post fenestram is an evagination of the periotic tissue into the cartilaginous capsule just posterior to the oval window. The fossula is also commonly involved by otosclerotic lesions.
The Pathology of Otosclerosis
ETIOLOGY
CLINICAL PRESENTATION
Age of Onset
Prevalence
Race
Gender
OTOSCLEROSIS AND PREGNANCY
DOES OTOSCLEROSIS PRESENT ONLY AS A CONDUCTIVE HEARING LOSS?
HISTOLOGIC OTOSCLEROSIS
SIMILAR DISORDERS
SITES OF INVOLVEMENT
ANATOMY OF THE OTIC CAPSULE
HISTOPATHOLOGY
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


