Chapter 24 Syndromic Medullary Thyroid Carcinoma
MEN 2A and MEN 2B
Introduction
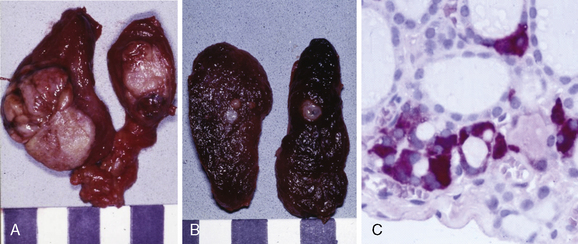
As early as the beginning of the 20th century, medullary carcinoma of the thyroid (MTC) was noted as a distinct disease entity within the broad spectrum of malignant thyroid tumors.1–4 By the first half of the century, the peculiar tumor biology of the sporadic type had been described as “small tumors with early lymphangic and hematogenous spread,”4 whereas multicentric MTC5 was being appreciated as the hallmark of familial tumors. Probably the first report of multiple endocrine neoplasia (MEN) 2B in a 12-year-old boy was given by Walther Burk in 1901.1 Yet it was E.D. Williams who clearly defined syndromic MTC as a separate entity in 1965 by publishing the first comprehensive series of hereditary pheochromocytoma (PCC) associated with MTC.6 The 1993 detection of the susceptibility gene of hereditary MTC by Mulligan et al.7 and Donis-Keller et al.,8 ushered in the era of genotype-phenotype oriented prophylactic surgery9–12 (Figure 24-1), completed a 100-year evolutionary saga of clinical, biochemical, and ultimately molecular understanding of hereditary MTC. Hereditary MTC, unique in many ways among hereditable cancer syndromes, is founded on the following findings (see also Chapters 23, Sporadic Medullary Thyroid Carcinoma, and 25, Sporadic Medullary Thyroid Microcarcinoma):
• The hereditary variant, affecting as many as 25% of MTC patients,13 is more frequent than many other common hereditary tumors.14
• Unlike many other hereditary tumors, hereditary MTC features a strong genotype-phenotype correlation12,15 that is utilized worldwide for risk assessment.16–18 This genotype-dependent, age-related tumor progression12 not only underlies the development of hereditary MTC but also the formation of MEN 2–associated pheochromocytoma (PCC)19 and hyperparathyroidism (HPT).20–23
• Disease progression from C-cell hyperplasia to MTC, requiring the acquisition of somatic mutations for malignant progression, is a stochastic sequence of events not fully under the control of a gene carrier’s genetic makeup.24 Serum calcitonin, a sensitive diagnostic marker of MTC, better reflects a gene carrier’s stage of C-cell disease than his or her underlying germline mutation in the RET (REarranged during Transfection) proto-oncogene.25 We may now tailor the timing and extent of surgical intervention in the neck to the gene carrier’s stage of disease through consideration of serum calcitonin levels.26
• Lymph node metastases are indicative of progressive disease, portending a worse prognosis for sporadic and hereditary MTC alike.27 Although multifocal tumor growth is more common in hereditary than sporadic MTC (65% versus 8%),28 no difference in biochemical cure and survival between hereditary and sporadic disease has been found after adjusting for extent of disease.27,29
• MEN 2B is a special and virulent variant of MEN type 2 characterized by the presence of MTC in early infancy. More than 90% of MEN 2B RET gene carriers harbor de novo germline mutations. Such de novo germline mutations are rare in other MTC syndromic settings such as familial MTC (FMTC) and MEN 2A.21,30 DNA-based screening of families for the MEN 2B trait for early detection of hereditary MTC hence is rarely an option. Intriguingly, there are some early clues in MEN 2B infants, notably “tearless crying” and “pseudo-Hirschsprung’s disease.” These clinical signs may help identify gene carriers before they develop the more characteristic MEN 2B stigmata, prompting rapid DNA-based screening and immediate surgical intervention.30
Clinical Presentation by Genotype
Familial Medullary Thyroid Cancer and MEN 2A
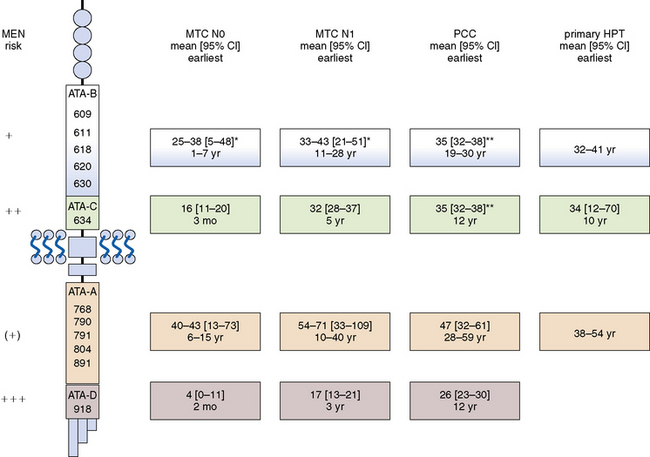
Activating germline mutations in the RET proto-oncogene on chromosome 10q11.2, which cluster in “hot spot” regions, by definition involve all cells of the body (also termed the first hit in Knudson’s model on oncogenesis)17,19,23–25,31 (see Figure 24-1). It should be noted that these germline mutations do not equally affect all neuroendocrine and nonendocrine tissues, and so many tissues may not be affected at all.24 To add another layer of complexity, the appearance of the various syndromic components is largely age dependent and characterized by a great deal of variability, especially among those gene carriers whose germline mutations have the weakest transforming activity. Different tissue-specific susceptibilities are the main driver behind FMTC/MEN 2A/MEN 2B phenotypes, all of which share MTC as an integral element.24
Familial Medullary Thyroid Cancer (FMTC)
Originally defined as an entity in its own right based on a minimum of four affected kindreds, FMTC is now conceived as an abortive form of MEN 2, which becomes manifest with the subsequent development of PCC or HPT. Such a conversion from an FMTC-only to a MEN 2 phenotype during the patient’s lifetime is the rule rather than the exception for RET germline mutations in codon 918 (American Thyroid Association [ATA] class D) and 634 (ATA class C), common for mutations in codons 620 and 618 but to a lesser extent for those in codons 611 and 609 (ATA class B), and rare for mutations in codons 768, 790, 791, 804, and 891 (ATA class A).18 As many as 9.5% of patients with seemingly sporadic MTC may harbor RET germline mutations.32 These gene carriers cannot be recognized unless all relevant exons of the gene have been screened.17,26
Multiple Endocrine Neoplasia Type 2A (MEN 2A)
The RET germline mutations underlying MEN 2A (Figure 24-2) involve mainly codon 634 (ATA class C) as the classic “MEN 2A codon,” infrequently codons 620, 618, and to a lesser degree codons 611 and 609 (ATA class B), and exceptionally codons 768, 790, 791, 804, or 891 (ATA class A).
Neuroendocrine Components in FMTC/MEN 2A
Medullary Thyroid Cancer (MTC)
Representing the usual first component of the MEN 2A syndrome, MTC is the only element common to FMTC, MEN 2A, and MEN 2B phenotypes. RET germline mutations are believed to drive C-cell hyperplasia, which can evolve to frank MTC with the acquisition of additional somatic mutations (“second hits”) in RET or other genes.12 C-cell hyperplasia often is multifocal, involving both thyroid lobes, especially in carriers of the stronger germline mutations in codon 918 (ATA class D) and 634 (ATA class C). Because it takes time to acquire those somatic mutations necessary for malignant transformation, the development of MTC is age-dependent and varies by affected RET codon18,24,25 (see Figure 24-2): ATA class D mutations: by the time of birth, ATA class C mutations as early as age 1 year, ATA class B mutations as early as age 5 years, and ATA class A mutations as early as age 10 years.
Pheochromocytoma (PCC)
Owing to the lower susceptibility of adrenal medullary relative to thyroidal C-cells, PCC represents the second most frequent syndromic manifestation.19 Among carriers with PCC, both conditions (i.e., MTC and PCC) coexist at the time of diagnosis in 60% with ATA class D (codon 918) and in 35% with ATA class C (codon 634) mutations. Only rarely is a solitary PCC the initial MEN 2 component among carriers with PCC: 0% with ATA class D (codon 918), 36% with ATA class C (codon 634), 40% with ATA class B (codon 620 or 618), and 100% with ATA class A mutations involving codon 791.19 Adrenal ganglioneuromas perhaps represent another, though unusual, component of the MEN 2A and 2B syndromes.33
Activating RET germline mutations are also thought to give rise to adrenal medullary hyperplasia, from which PCC may develop upon allelic loss at chromosome 1p or deletion of the von Hippel-Lindau gene. This process varies by affected codon19 (see Figure 24-2): ATA class D and C mutations: as early as age 10 years, ATA class B and A mutations: as early as age 20 years.
Hyperparathyroidism (HPT)
Parathyroid chief cells may not be as responsive to RET activation as parafollicular C-cells of the thyroid or adrenal medullary cells. For unknown reasons, HPT, which is generally mild, is not an integral part of the MEN 2B syndrome. It is fairly common in older carriers of RET germline mutations in codon 634 (ATA class C)23 but infrequent in carriers of weaker activating RET mutations in codons 620, 618, 611, and 609 (ATA class B) or 768, 790, 791, 804, or 891 (ATA class A). Because of the infrequency of HPT, little information is available for carriers of RET germline mutations in codons other than 634. Again, the development of HPT is age-dependent (i.e., to acquire “second hits”) and varies by affected codon (see Figure 24-2): ATA class C mutations as early as age 10 years, ATA class B mutations as early as age 30 years, and ATA class A mutations with no estimate owing to the current paucity of data.
Nonendocrine Components in FMTC/MEN 2A
Hirschsprung’s (HSCR) Disease
Intriguingly, 6% to 16% of RET families with ATA class B germline mutations in exon 10 (codons 620 and 618, and more rarely 611 and 609) develop a peculiar phenotype consisting of both a “gain of function” FMTC/MEN 2A phenotype and a “loss of function” HSCR phenotype.34 These genotypes have been dubbed “Janus genes,” referring to the Roman god of doorways facing either way. Occasional reports have also described instances of HSCR disease in carriers of RET germline mutations in codon 791.35 Frequently, HSCR disease precedes the clinical manifestation of MTC and PCC. The “loss of function” HSCR phenotype is thought to be due to the trapping of precursor RET receptor proteins in the endoplasmatic reticulum, precluding their transport to the cell membrane. Decreased cell surface concentrations of RET receptor protein are believed to cause the premature arrest of the craniocaudal migration of enteric neurons during the 5th to 12th week of gestation. The absence of autonomic ganglion cells (aganglionosis) within parasympathetic submucosal and myenteric plexuses causes the HSCR symptoms of functional obstruction and megacolon.34 In stark contrast, gastrointestinal ganglioneuromatosis, also dubbed “pseudo-Hirschsprung’s disease, which is a feature of MEN 2B (codon 918), is characterized by the presence of ganglioneuromas in these plexus.
Cutaneous Lichen Amyloidosis (CLA)
This condition may occur in as many as 9% (18 of 199) of MEN 2A families.22 It is almost always associated with RET germline mutations in codon 634, with one reported instance of a RET germline mutation in codon 804.36 Itching is the usual first manifestation of CLA. It is typically located in the upper back at either or both interscapular regions in an area of repeated scratching, involving the dermatomes between C4 and T5.37 The neural crest cells, implicated in the embryonic development of the parafollicular C-cells and adrenal medulla, are also involved in the embryogenesis of the thoracic sensory fibers. This may explain the frequent observation that these dermatologic symptoms often precede the clinical manifestation of MTC.36 Yet the evolution of CLA is independent of the course of the disease and unrelated to the development of PCC or HPT.37
MEN 2B
MEN 2B is defined by the combination of MTC and PCC, the absence of HPT, and the presence of ocular, oral, intestinal, and musculoskeletal ganglioneuromas and unique musculoskeletal disorders.18,22 MTC develops so early that it is (1) present in virtually every MEN 2B patient, (2) frequently advanced at the time of diagnosis, and (3) associated with a bleaker prognosis. PCC are expressed in more than 50% of patients frequently involving both adrenals.18,22
Nonendocrine Components in MEN 2B
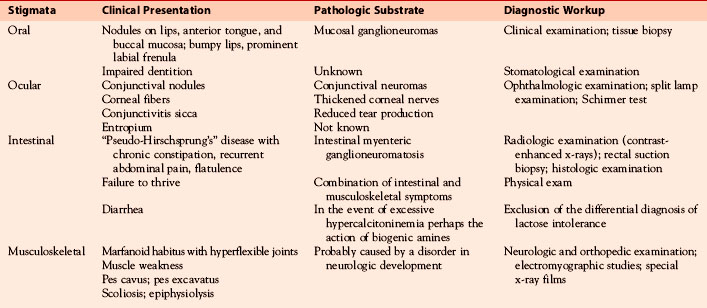
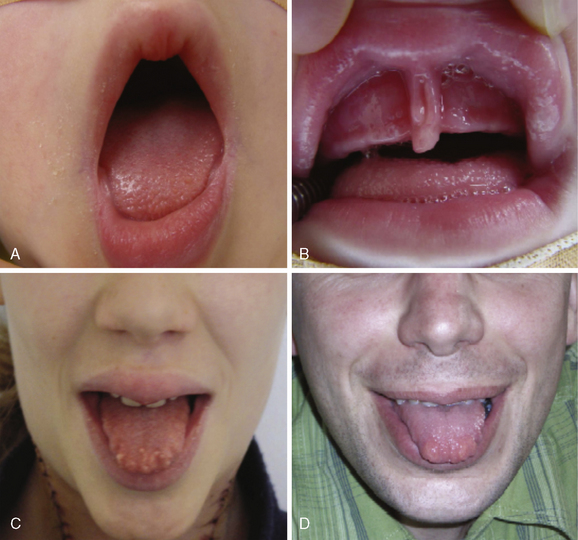
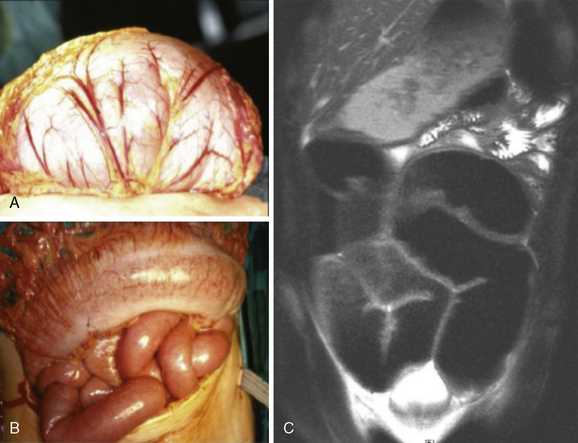
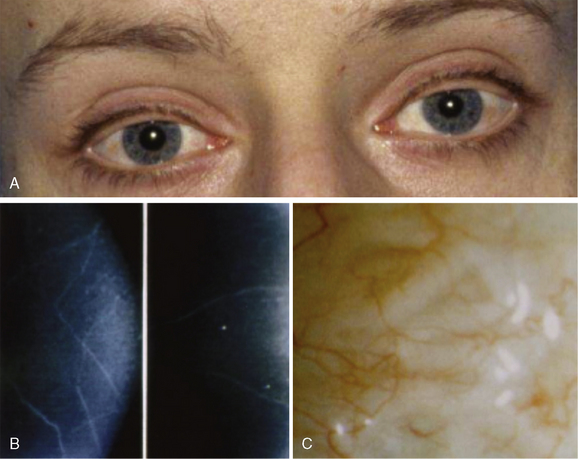
The nonendocrine components encompass oral, ocular, intestinal, and musculoskeletal stigmata (Table 24-1, Figures 24-3 to 24-5). The oral stigmata include nodules of the anterior tongue, lips and buccal mucosa,38,39 as well as dental irregularities.40 The ocular stigmata comprise conjunctival nodules with recurrent conjunctivitis, thickened corneal nerves (corneal fibers), and infolding of the eyelid margins (i.e., entropium).38,41–44 The intestinal ganglioneuromatosis usually presents as chronic constipation or bowel obstruction.45–48 The musculoskeletal stigmata consist of a variety of distinct malformations such as hyperflexible joints, muscle weakness, midface hypergnathism, pectus deformities, scoliosis, slipped capitis femoral epiphysis, and feet abnormalities. The characteristic elongated extremities are reminiscent of a “marfanoid habitus.”49,50
Although the syndrome was described (at least in parts) much earlier,1,41,51–54 the term MEN 2B was introduced only in 1975 to signify this “neuroma phenotype” from the classic Sipple’s syndrome.55 Subsequent series disclosed a large variability in the expression of the endocrine and nonendocrine MEN 2B components.39,56–58
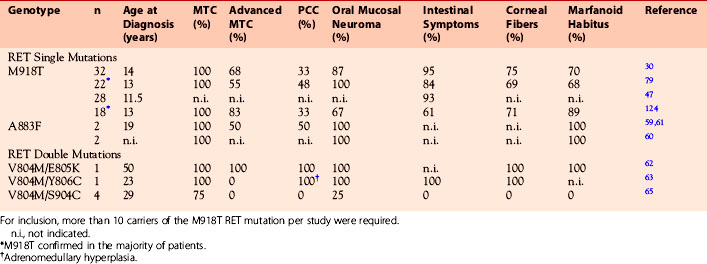
In fewer than 5% of MEN 2B patients who may not express the nonendocrine phenotype in full (“atypical MEN 2B”), other RET germline mutations, including double mutations, may be present, including A883F,59–61 V804M/E805K,62 V804M/Y806C,63,64 and V804M/S904C65 (see Table 24-2). These atypical MEN 2B mutations tend to be less aggressive and less penetrant than the classic MEN 2B mutation in codon 918 (ATA class D), which is consistent with in vitro studies.66,67
The endocrine and nonendocrine phenotypes of MEN 2B develop in an age-dependent fashion30,68 (Table 24-3; also see Figure 24-3). Carriers of the classic MEN 2B mutation in codon 918 frequently harbor MTC in the first months of life48,69 when surgical cure is still within reach. This window of opportunity closes rapidly within the first years of life so that most MEN 2B patients who are older than four years at the time of diagnosis are not curable.68 PCC as the second part of the endocrine phenotype in MEN 2B is rarely seen before puberty,19 the youngest MEN 2B patient reported being 12 years of age.58
Table 24-3 Age-Dependent Development of Extrathyroidal MEN 2B Stigmata and Symptoms (M918T Mutation) Relative to Healthy Controls within the First Year of Life
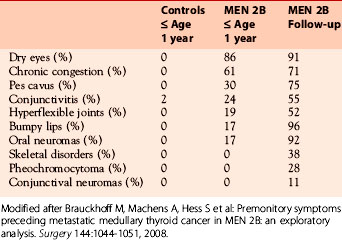
The majority of carriers of the classic RET mutation in codon 918 undergo an uneventful intrauterine development and birth, demonstrating normal body weight and size. No pertinent data are available for the atypical MEN 2B mutations. Some months after birth, failure to thrive due to suckling weakness and chronic constipation may give the first clue of MEN 2B.30
Ganglioneuroma and thickened nerve sheaths probably are at the heart of the nonendocrine MEN 2B components, pointing to the RET receptor’s key role in neural crest development.70 Prominent corneal nerves, which are visible on split lamp examination, and thickened recurrent laryngeal nerves (Figures 24-5 and 24-6) are common. The neural findings are caused by thickened nerve sheaths, which reduce nerve conduction velocity and action potential amplitudes within the Aa, Ad, and C components of sensory nerves.49 This pathomorphology likely is the driver not only behind the musculoskeletal stigmata but also behind conjunctivitis and reduced tear production. Ganglioneuromas are expressed ubiquitously but abound in the oral (see Figure 24-3) and intestinal mucosa (see Figure 24-4) and the conjunctiva (see Figure 24-5). In the colon, ganglioneuromatosis interferes with gut motility and digestion, giving rise to “pseudo-Hirschsprung’s disease” 45 (see Figure 24-4).
Evaluation of Gene Carriers for Surgery
FMTC and MEN 2A-MTC
MTC cells, given their neuroendocrine derivation from the neural crest, synthesize calcitonin. This hormone is stored in dense cored secretory granules to be released into the bloodstream upon stimulation with provocative agents. Because intravenous injection of pentagastrin, typically 0.5 μg per kilogram body weight, can cause bothersome side effects, the use of pentagastrin has become restricted in some countries including the United States. Before the dawn of the molecular era, kindreds from clinically established MEN 2 families periodically underwent calcitonin screening with pentagastrin stimulation for early identification of the trait and, if positive, were referred for preemptive total thyroidectomy. Owing to the presence of nonneoplastic (“reactive”) C-cell hyperplasia, biochemical screening occasionally yielded false-positive findings prompting unnecessary thyroidectomies in family members who subsequently were shown not to have inherited the RET germline mutation running in that family. Naturally, the risk of harboring occult MTC is greater the higher are these calcitonin levels. Calcitonin screening, being a powerful diagnostic tool, has greatly diminished the excess mortality of RET gene carriers (typically this was due to hypertensive crisis [catecholamine excess] from an unrecognized PCC or to metastatic MTC).71
With the advent of the molecular age, DNA-based screening of the “hot spot” regions in exons 10, 11, 13, 14, 15, and 16 of the RET gene has quickly become the diagnostic gold standard. Barring sample/lab error, there are no false-positive findings with DNA-based screening, which has largely replaced calcitonin screening for early diagnosis of gene carriers.17 Nevertheless, increased calcitonin levels are a more sensitive marker of occult MTC than high-resolution ultrasonography, which has great difficulty in detecting tumors smaller than 5 millimeters, especially before a backdrop of benign nodular disease.72
Pheochromocytoma (PCC)
MEN 2–associated PCC, displaying impairment in terminal differentiation, frequently produce epinephrine rather than norepinephrine.73 By implication, many RET gene carriers with PCC sustain paroxysmal rather than sustained hypertension. When a delay in diagnosis allows PCC to progress, minor triggers can provoke excessive catecholamine secretion igniting an often lethal hypertensive crisis, typically around the fourth to fifth decade. In former times, this used to be the most common cause of death among RET gene carriers. With the routine use of sensitive screening tests for catecholamines and their metabolites, PCC are increasingly being diagnosed at a yet asymptomatic stage.74 These adrenal medullary tumors usually are large enough to visualize on adrenal ultrasonography, computed tomography, and magnetic resonance imaging. To circumvent the risk of sparking a hypertensive crisis, no manipulation of these tumors must be undertaken before an effective alpha-receptor blockade has been put in place.75
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


