Syndromes
Alex V. Levin
Thomas W. Wilson
Elise Héon
The eye is the second most commonly involved organ in systemic syndromes after the central nervous system. Most syndromes are genetic in origin with the expression of abnormal genes in various tissues throughout the body. Multisystem involvement may occur because the same gene plays a vital role in each affected tissue or because more than one gene is involved, as in a contiguous gene deletion syndrome. Patients may present with ocular abnormalities that lead to the diagnosis of an underlying syndrome. Knowing the ocular manifestations of syndromes also allows for appropriate ophthalmic screening.
The number of known syndromes is in the thousands and is far beyond the scope of this chapter and the knowledge base of any one ophthalmic practitioner. Perhaps the most comprehensive source of information can be found in the online version of Mendelian Inheritance in Man (http://www-ncbi-nlm-nih-gov.easyaccess1.lib.cuhk.edu.hk/omim/). Syndromes with a major manifestation in a particular organ system may be found elsewhere in this atlas. For example, Apert syndrome is discussed in Chapter 14: Craniofacial; WAGR syndrome in Chapter 6: Iris and Pupils; and Alagille syndrome in Chapter 17: Gastrointestinal.
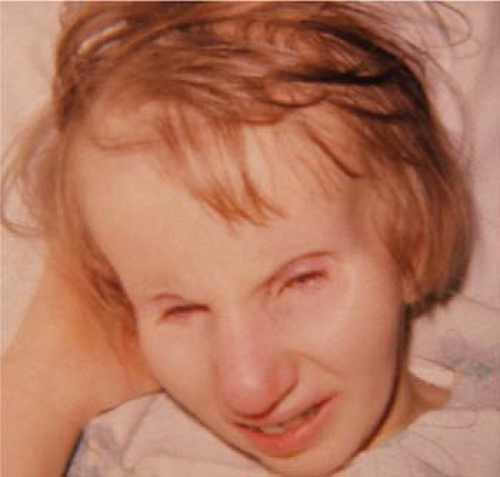 Figure 29.1 Cockayne Syndrome Cockayne syndrome is an autosomal recessive disorder characterized by rapid premature aging, deafness, miosis, cataract, and pigmentary retinopathy similar to retinitis pigmentosa (Chapter 8: Retina and Vitreous). Microphthalmia may also occur. Facial features include microcephaly with loss of facial adipose tissue and dermatitis secondary to sun exposure, which can also be seen in combination with xeroderma pigmentosa (Chapter 15: Dermatology, Fig. 15.6). Patients typically have some degree of developmental delay and growth retardation. Fibroblast cultures from patients with Cockayne syndrome have an increased sensitivity to ultraviolet light, and the underlying mechanism is most likely secondary to an inability to repair the skin following exposure to the sun. Patients with Cockayne syndrome can have pigmentary degeneration of the retina. |
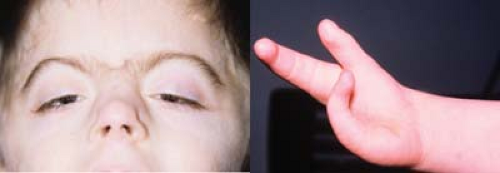 Figure 29.2 Cornelia de Lange Syndrome (Brachmann-de Lange Syndrome) Cornelia de Lange syndrome is a genetic abnormality due to mutations in the NIPBL gene at 5p13.1 in 50% of patients, with a characteristic facies, variable mental and growth retardation, skeletal abnormalities ranging from small hands to severe limb reduction, and gastroesophageal reflux. Other findings may include deafness, cardiac abnormalities, Raynaud phenomena (Chapter 27: Rheumatology Fig. 27.18), and cleft palate. The most common facial features include hypertrichosis of the eyebrows with a “V shaped” synophrys and long arcuate eyelashes. Other ocular manifestations include unilateral or bilateral congenital ptosis, myopia, peripapillary hyperpigmentation, and blepharitis. Nasolacrimal anomalies and nystagmus may also be seen. |
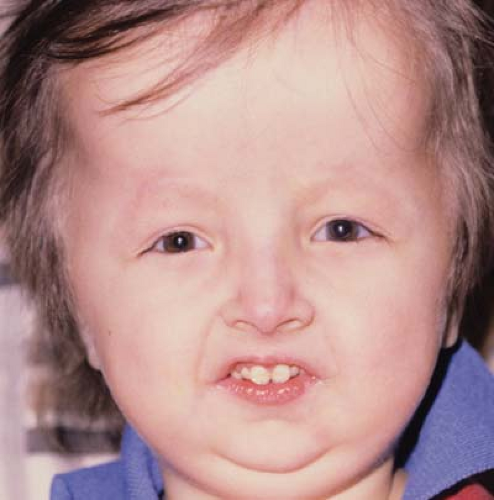 Figure 29.3 Hallermann-Streiff Syndrome (Oculomandibulodyscephaly with Hypotrichosis Syndrome)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|