Site of primary airway anomaly
Syndrome
Nasal Cavity and Midface
Congenital nasal pyriform aperture stenosis
CHARGE syndrome
Craniosynostosis syndromes (Crouzon, Apert, Pfeiffer)
Oromandibular
Stickler syndrome
Treacher Collins syndrome
Craniofacial microsomia
Laryngotracheal
Complete tracheal rings in Down syndrome (Trisomy 21)
Tracheal cartilaginous sleeve in craniosynostosis syndromes
Table 2
Specific airway concerns with craniofacial syndromes
Craniofacial syndrome | Specific airway concerns |
|---|---|
Congenital nasal pyriform aperture stenosis | Narrow pyriform aperture width |
Can have associated narrow nasal cavities | |
CHARGE syndrome | Unilateral choanal atresia |
Bilateral choanal atresia | |
Laryngomalacia | |
Pharyngolaryngeal hypotonia | |
Craniosynostosis syndromes | Midface hypoplasia—nasopharyngeal and oropharyngeal obstruction |
Obstructive sleep apnea | |
Nasal cavity stenosis | |
Choanal atresia | |
Tracheal anomalies—tracheal cartilaginous sleeve | |
Stickler syndrome | Macroglossia |
Micrognathia | |
Treacher Collins syndrome | Severe micrognathia |
Unilateral choanal atresia | |
Bilateral choanal atresia | |
Obstructive sleep apnea | |
Craniofacial microsomia | Mandibular hypoplasia |
Down syndrome | Complete tracheal rings resulting in tracheal stenosis |
Subglottic stenosis | |
Laryngomalacia | |
Tracheobronchomalacia | |
Macroglossia | |
Midfacial hypoplasia | |
Obstructive sleep apnea | |
Pharyngeal hypotonia |
Nasal Cavity and Midface
Nasal obstruction can cause respiratory distress immediately after birth in the neonate who is an obligate nasal breather. It has been suggested that this phenomenon is due to the prominent soft palate and relatively higher epiglottis in the neonate with a resultant smaller oropharyngeal airway. The neonate can have cyclical breathing in which he/she initially has nasal obstruction, develops cyanosis and hypercapnia, and then begins to cry and ventilate orally. When the cyanosis subsides, the neonate calms and attempts to breathe nasally, thus starting the cycle over again. These cyanotic episodes and ineffective ventilation can result in death if not discovered quickly. Neonates gradually outgrow this sole reliance on nasal breathing by about 4–6 months of age. Obstruction at the nasal level is commonly due to nasal pyriform aperture stenosis, choanal stenosis or atresia, and midface hypoplasia.
Congenital Nasal Pyriform Aperture Stenosis
Etiology. Congenital nasal pyriform aperture stenosis (CNPAS), first described in 1989, is believed to occur when bony overgrowth of the nasal processes of the maxilla narrows or obstructs the nasal cavities [12]. CNPAS can occur in isolation, but there are frequently additional findings suggestive of a microform holoprosencephaly (HPE). The full spectrum of HPE has been linked to many chromosomal abnormalities.
Epidemiology. The true incidence of CNPAS is unknown but occurs less commonly than choanal stenosis or atresia.
Pathogenesis. HPE, the abnormal division of the forebrain, varies widely in severity. A single central maxillary incisor, present in approximately 60 % of CNPAS, is considered the least severe form of HPE [5, 43]. Additional findings linked with CNPAS can include abnormal development of the brain and the pituitary gland leading to endocrine dysfunction.
Clinical presentation. CNPAS causes symptoms of nasal obstruction, which develop shortly after birth. In mild cases, the patient may present with “noisy breathing,” aggravated with feeding and agitation. In severe cases, cyclical breathing may be present with cyanotic episodes.
Diagnosis. The inability to pass a suction catheter larger than 5 Fr through the anterior nares is suspicious for CNPAS and should be confirmed by an otolaryngologist with nasal endoscopy. CT imaging can help evaluate for other contemporaneous midline defects such as a single central maxillary incisor (Fig. 1). A pyriform aperture width, as viewed on CT axial imaging, of less than 11 mm in a term infant is considered to be diagnostic for CNPAS [7]. Careful evaluation is necessary to distinguish CNPAS from choanal atresia, which can present in a similar fashion.
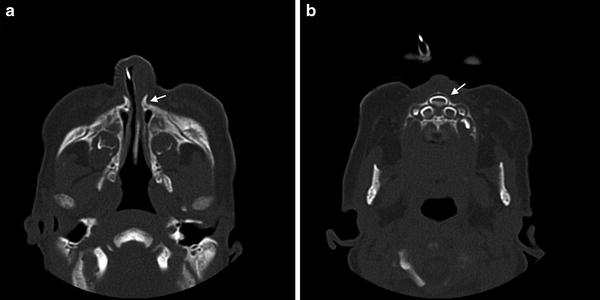
Fig. 1
Congenital nasal pyriform aperture stenosis (CNPAS). (a) Axial computed tomographic (CT) image showing a narrow 4 mm pyriform aperture width. (b) Concurrent single central maxillary incisor
Management. Neonates with CNPAS often require airway monitoring in the intensive care unit. Conservative management, including support with special feeding techniques and monitoring until the airway grows, is indicated in mildly symptomatic neonates. Nasal decongestants, humidification, and nasal continuous positive airway pressure (CPAP) may be sufficient. Surgery to widen the nasal pyriform aperture, accessed through a sublabial incision, is often required in the more severely afflicted neonates. Some feel that CNPAS is associated with an overall narrowing of the width of bilateral nasal cavities. Thus, dilation of the nasal cavities with inferior turbinate outfracturing may be required with possible temporary nasal stent placement.
Multidisciplinary considerations. A brain MRI and blood tests for pituitary hormone levels should be considered to assess for pituitary dysfunction. The need for consultation of specialists, such as endocrinology and neurology, depends on the constellation of HPE-associated findings. Developmental delay of variable severity may occur in children with HPE and patients should be followed long-term.
CHARGE Syndrome
Etiology. The CHARGE acronym stands for Coloboma of the eye, Heart defects, Atresia of the choanae, Restricted growth, Genital anomalies, and Ear anomalies. CHARGE syndrome, also known as Hall Hittner syndrome, was first described in 1979. CHARGE is associated with mutations in the CHD7 gene, but approximately one-third of cases have no identified mutation [24]. Most cases arise de novo and represent a single occurrence in a family, but an autosomal dominant inheritance pattern is seen in familial CHARGE syndrome.
Pathogenesis. CHD7 gene mutations affect the protein involved in chromatin remodeling, which disrupts the regulation of gene expression [13].
Clinical presentation. Choanal stenosis or atresia can cause life-threatening respiratory distress in neonates (Fig. 2). Unilateral or bilateral choanal atresia is present in 50–60 % of patients [24]. Laryngomalacia is a major cause of upper airway obstruction and can be present in 8–37 % of patients with CHARGE [27]. Pharyngolaryngeal hypotonia can result in additional upper airway obstruction. Patients may also have cranial neuropathies involving cranial nerves V, VII, VIII, IX, and X [27]. Cardiovascular instability and feeding issues such as dysphagia and aspiration can present additional significant challenges in the neonatal period. As many as 80 % of children with CHARGE may have gastroesophageal reflux disease [27], which can further exacerbate airway obstruction.
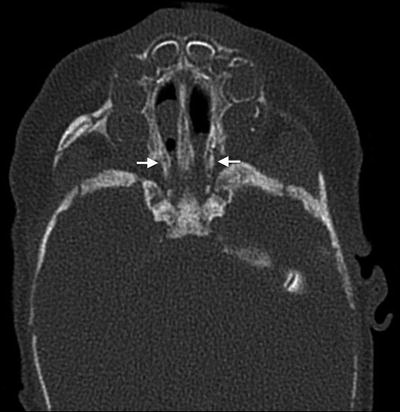
Fig. 2
Bilateral mixed membranous and bony choanal atresia in a patient with CHARGE syndrome
Diagnosis. Mutations in the CHD7 gene are identified by genetic testing in approximately 67 % of individuals with a phenotype consistent with CHARGE [38]. Four major defining characteristics in CHARGE are ocular coloboma, choanal stenosis or atresia, cranial nerve dysfunction (varies from dysphagia, sensorineural hearing loss, hyposmia, or facial palsy), and anomalies of the middle or inner ear. The minor characteristics that are less specific but commonly seen in CHARGE are cardiovascular anomalies, growth delays, developmental delay and genital hypoplasia, which are more apparent in males. A clinical diagnosis can be made if an individual has four major characteristics or three major with one minor characteristic [10].
Management. Airway and cardiac evaluations are required immediately. Airway management may require intubation, transnasal or transpalatal surgical repair of choanal stenosis or atresia, or tracheotomy. A tracheotomy may need to be performed in as many as 10–60 % of patients [27]. A supraglottoplasty for management of severe laryngomalacia should be performed only after a thorough airway evaluation as children with CHARGE syndrome have poorer outcomes likely due to concurrent airway pathology. Feeding difficulties can be a major source of morbidity requiring speech and swallow therapy.
Multidisciplinary considerations. Specialist consultations of cardiology, otolaryngology, ophthalmology, audiology, and speech pathology services are usually needed. Individuals with CHARGE usually have feeding issues and variable learning disabilities worsened by dual visual and hearing impairment. Serial audiologic and ophthalmologic exams are recommended. Hypogonadism may disrupt progression into puberty.
Craniosynostosis Syndromes
Etiology. Mutations in the fibroblast growth factor receptor (FGFR) genes account for many craniosynostosis syndromes including Crouzon, Apert and Pfeiffer syndromes. FGFR genes encode different fibroblast growth factor receptors that regulate cell growth and embryonic development. The FGFR mutations are transmitted in an autosomal dominant inheritance pattern [36].
Epidemiology. Crouzon syndrome affects 16 per million newborns [15]. Apert syndrome affects between 1:65,000 and 1:88,000 newborns [3], and Pfeiffer syndrome affects 1:100,000 newborns [45].
Pathogenesis. Mutations in the FGFR protein affect the development of bone cells and cause premature fusion of the sutures of the skull (Fig. 3). Craniosynostosis can cause airway obstruction from midface hypoplasia or tracheal abnormalities.
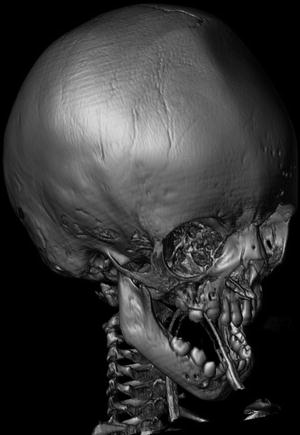
Fig. 3
Patient with Crouzon syndrome with premature fusion of the cranial sutures. Three-dimensional reconstructed image from CT images shows ridge in the midline of the frontal bone which results from early fusion of the metopic suture
Clinical presentation. An abnormally shaped skull is apparent at birth in a neonate with craniosynostosis. The degree of airway compromise secondary to midface hypoplasia is variable, and almost half of children with Crouzon, Apert and Pfeiffer syndromes develop obstructive sleep apnea [6]. Nasal cavity stenosis or choanal atresia may also be present in this population. Additionally, neonates with Crouzon, Apert and Pfeiffer syndromes may have stridor and respiratory distress secondary to tracheal abnormalities, which are discussed later in this chapter. In one large retrospective review, children with craniofacial synostosis syndromes had the highest rate of tracheotomy at 48 % [39].
Diagnosis. Genetic testing is available, but a clinical diagnosis is frequently possible for most of the FGFR-related craniosynostosis syndromes based on facial features, distinguishing anomalies of the hands and feet, and the cranial sutures affected. These three craniosynostosis syndromes share common features of maxillary hypoplasia, shallow orbits causing proptosis, wide set eyes and a beaked nose. Apert syndrome is notable for syndactyly of the fingers and toes whereas normal hands and feet are typical for Crouzon syndrome [4, 16, 35]. Individuals with Pfeiffer syndrome tend to have partial syndactyly and medially displaced, broad and short thumbs and big toes [30, 45].
Management. Expansion of the cranial vault and midface advancement require staged surgeries. Tracheotomy may be needed for airway management especially when multiple surgeries are often needed to address the craniofacial abnormalities. Midface advancement can help alleviate obstruction of the nasopharyngeal and oropharyngeal airway. Choanal atresia repair or nasal splints may be needed to address nasal obstruction. Adenotonsillectomy may partially alleviate OSA if hypertrophy of these structures develops later in childhood.
Multidisciplinary considerations. Long-term care should be managed by a craniofacial team involving neurosurgery, plastic surgery, otolaryngology, ophthalmology, genetics, and developmental pediatrics. Intellectual disability affects individuals in certain types of FGFR-related craniosynostosis. Cognitive function is normal in Crouzon syndrome, but variable intellectual disability is common in Apert and Pfeiffer syndromes [23]. Individuals with craniosynostosis may require lifelong monitoring for hydrocephalus.
Oromandibular
An underdeveloped mandible can cause upper airway obstruction due to the resulting posterior or downward displacement of the tongue.
Stickler Syndrome
Etiology. Stickler syndrome is a group of connective tissue disorders characterized by distinctive orofacial features, hearing loss, premature degenerative joint disease, progressive myopia, and retinal detachment [41]. The subtypes of Stickler syndrome (types I through V) are associated with mutations in the genes COL2A1, COL11A1, COL11A2, COL9A1, and COL9A2, respectively, with types I through III being most common [41]. The inheritance pattern could be either autosomal dominant or recessive depending on the subtype.
Epidemiology. The estimated incidence of Stickler syndrome is between 1:7,500 and 1:9,000 [35]. Approximately 35 % of newborns with Pierre Robin sequence develop features of Stickler syndrome.
Pathogenesis. Mutations in any of these genes affect the production of collagen types II, IX, and XI. Connective tissues containing these collagen fibers do not develop normally.
Clinical presentation. Common orofacial findings include the Pierre Robin sequence of macroglossia, cleft palate, and micrognathia (Fig. 4). Some neonates may present with airway distress requiring intubation in severe cases of micrognathia. Cleft palate occurs in about 40 % and causes feeding difficulties [37]. Additional facial characteristics, such as a hypoplastic flattened midface with a depressed nasal bridge, are more prominent in younger children than in adults [40]. Hearing loss affects approximately 63 % of individuals and is predominantly sensorineural (67.8 %) from abnormal collagen in the inner ear [1]. A mixed or conductive hearing loss is common in individuals with a cleft palate from chronic Eustachian tube dysfunction. Common eye problems include myopia and vitreous abnormalities. The different subtypes of Stickler syndrome have specific associated features for each genotype, but phenotypic variability is common even within families with the same genotype.
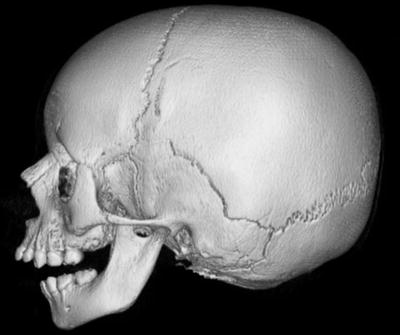
Fig. 4
Patient with Pierre Robin sequence. Three-dimensional reconstructed image from CT images shows retrognathia
Diagnosis. Genetic testing is available, but the diagnosis is considered when this combination of orofacial, ocular, auditory, and musculoskeletal manifestations are present.
Management. The feeding and airway problems present in the neonatal period often require care coordination to determine the timing of cleft palate repair and airway management. Airway management is aimed at relieving base of tongue obstruction at the level of the oropharynx. Non-operative management includes prone positioning, oral airway placement, nasopharyngeal stenting, and short-term intubation. Operative management includes tongue-lip adhesion and mandibular distraction osteogenesis [34]. Tracheotomy is reserved for severe airway obstruction.
Multidisciplinary considerations. Long-term care should be coordinated by a craniofacial team including plastic surgery, oromaxillofacial surgery, otolaryngology, and genetics. Children with Stickler syndrome should have an eye exam performed annually by a vitreoretinal specialist and a hearing test biannually until age five then annually thereafter [35]. Screening for mitral valve prolapse should be performed during routine physicals. Contact sports should be avoided due to the underlying risk of retinal detachment.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


