Purpose
To evaluate the spectrum of morphologic abnormalities in patients with Wagner syndrome by spectral-domain optical coherence tomography (SD OCT).
Design
Retrospective comparative case study.
Methods
Institutional study of patients entered into the French Vitreoretinopathy Study Group database. Twelve eyes of 9 patients from 3 unrelated families with genetically confirmed Wagner syndrome and 28 eyes from 15 age- and sex-matched healthy family controls were scanned by SD OCT. Morphology and layer thickness of the total retina, inner retinal layers, outer retinal layers, and photoreceptor layer at different degrees of eccentricity from the fovea were compared between the 2 groups.
Results
A thick multilayered membrane adherent to the perifovea but completely detached from the fovea, thus forming a bridge over the foveal pit, was observed in 84% of eyes from patients with Wagner syndrome. At the equatorial area, SD OCT imaging allowed visualization of the architecture of an avascular vitreous veil with localized retinal traction. Most retinal layers were significantly thinner in patients with Wagner syndrome compared to the control group, except at the foveal center where abnormal persistence of 1 or more inner retinal layers could be observed.
Conclusion
SD OCT provides better structural insight into the range of retinal defects at the vitreoretinal interface and fovea, which is not only useful for improving diagnosis and management, but also for understanding the pathogenesis of Wagner syndrome.
Wagner syndrome (OMIM#143200) is a very rare vitreoretinal degenerative disorder with no systemic features, inherited as an autosomal dominant trait, and only 14 families have been reported worldwide with a molecularly confirmed diagnosis. This genetic disorder is caused by splice mutations in the versican ( VCAN ) gene, coding for a chondroitin sulfate proteoglycan named versican whose exact role in ocular tissues is largely unknown. The pattern of expression of versican during retinal development and in the adult retina supports a role in vitreous architecture and vitreoretinal interface, as well as in the maintenance of photoreceptor cells, and in the regulation of neurite formation and growth of the nerve fiber layer and inner plexiform layer where neural networks of ganglion cells are being formed. The clinical spectrum of the disease is very large and age-dependent, with a highly variable expressivity even among affected members within a same family, making diagnosis of this disease very challenging. The disease usually manifests in childhood, and affected patients show an optically empty vitreous with a characteristic fibrillary aspect of the vitreous core and an abnormal vitreous cortex with avascular peripheral veils. Other features variably include moderate congenital myopia, early-onset cataract, glaucoma, uveitis, and foveal ectopia responsible for pseudostrabismus. The severity of the disease is related to the progressive chorioretinal degeneration with atrophy and the occurrence of retinal detachments, both of these complications being the leading causes of visual loss in patients with Wagner syndrome. Although retinal detachment has long been an unrecognized manifestation and is not as frequent in Wagner syndrome as in Stickler, all recent studies have reported retinal detachment in affected patients. Visual acuity is reported to be normal or subnormal in young patients with Wagner syndrome but is constantly severely affected in older patients.
To gain insight into the qualitative and quantitative defects in the retina and at the vitreoretinal interface in Wagner syndrome, a retrospective intrafamilial comparative case study was conducted using the in vivo spectral-domain optical coherence tomography (SD OCT) imaging method. The retinal structural information provided by SD OCT may help in clarifying the underlying pathologic mechanisms at play in Wagner syndrome, which remain poorly understood thus far.
Methods
Institutional review board approvals for retrospective chart reviews were obtained commensurate with the respective institutional requirements prior to the beginning of the study. Described research was approved by the Ethics Committee of the French Society of Ophthalmology and adhered to the tenets of the Declaration of Helsinki. Fully written informed consent was obtained for all patients.
This intrafamilial case-control study included patients followed for hereditary vitreoretinal diseases at a single institution, a university teaching hospital at Groupe Hospitalier Cochin Hôtel-Dieu, Paris, France. Patients enrolled in our cohort (The French Vitreoretinopathy Study Group) were offered standardized ophthalmic examination and genetic testing.
Patients with a clinical picture of Wagner syndrome and positive for a pathogenic mutation in the VCAN gene were enrolled in this study (cases). The control population included clinically unaffected family members who were non-carriers of VCAN mutations (controls). ETDRS best-corrected visual acuity (BCVA), slit-lamp biomicroscopy, intraocular pressure measurements, dilated fundus examination, and spherical equivalent and axial length measurement (IOLMaster; Carl Zeiss Meditec AG, Jena, Germany) was performed for all studied patients (cases and controls) and findings have been partly reported elsewhere ( Table 1 ). SD OCT was performed using the Spectralis OCT (Heidelberg Engineering, Heidelberg, Germany). Patients with no SD OCT data; with a history of retinal detachment, macular edema, or premature birth; or with low-quality SD OCT images were excluded from the analysis (see Figure 1 for inclusion flow chart details).
| Family ID | VCAN Mutation | References | Patient ID | Sex | Age (y) | Eye | BCVA (LogMAR) | AL (mm) | SE (Diopters) | IOP | ExRM | Grade 1 FH | Grade 2 FH |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | c.4004-2A>T | Brézin et al | A.IV.7 | M | 61.9 | OD | 0.4 | 25.05 | 17 | Yes | Yes | Yes | |
| A.IV.8 | M | 51 | OS | 0.1 | 24.77 | −7.75 | 11 | Yes | Yes | No | |||
| A.V.5 | F | 35.9 | OD | 0.1 | 23.84 | −3.375 | 16 | Yes | No | No | |||
| OS | 0.1 | 24.1 | −5.5 | 16 | Yes | No | No | ||||||
| A.V.6 | F | 40.7 | OD | 0.2 | 27.05 | −5.625 | 16 | Yes | No | No | |||
| B | c.4004-6A>T | Rothschild et al | B.I.2 | F | 72.6 | OS | 0.2 | 17 | Yes | No | No | ||
| B.II.1 | F | 44.7 | OD | 0.2 | 21.97 | 1 | 17 | Yes | No | No | |||
| B.II.4 | F | 47.4 | OD | 0.1 | −0.875 | 16 | Yes | No | No | ||||
| OS | 0.1 | −0.75 | 17 | Yes | Yes | No | |||||||
| C | c.9265+1G>A | Rothschild et al | C.II.1 | M | 30.5 | OS | 0 | 26.7 | −3.125 | 17 | Yes | Yes | Yes |
| C.III.1 | F | 3 | OD | 0.5 | −3 | No | Yes | No | |||||
| OS | 0.5 | −6.375 | No | Yes | No |
Spectral-Domain Optical Coherence Tomography Analysis
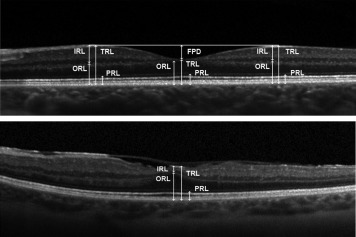
The SD OCT acquisition protocol included a horizontal and vertical 9-mm central linear scan (passing through the fovea). At least 30 scans were averaged to reduce the signal-to-noise ratio and low-quality images (signal-to-noise ratio below 25 dB) were discarded. Qualitative OCT image analysis included the quoting of the presence or absence of a membrane-like structure forming a bridge over the foveal pit according to our previous description in time-domain OCT of Wagner syndrome patients. We also graded foveal abnormalities as previously described by others. In brief, we noted the absence of extrusion of the plexiform layers (grade 1) from the central fovea, the absence of foveal pit (grade 2), the absence of outer segment lengthening (grade 3), and finally the absence of widening of the outer nuclear layer (grade 4). Quantitative analysis included manual segmentation of several retinal layers using the built-in caliper of the viewing software (HRA Spectralis viewing software version 5.6.4.0). We defined the following layers: (1) total retinal thickness (TRT), as the distance from the inner limiting membrane (ILM) to the inner aspect of the Bruch membrane (BM); (2) inner retinal layers (IRL), as the distance from the ILM to the outer plexiform layer (not included); (3) outer retinal layers (ORL), as the distance from the inner aspect of BM to the outer plexiform layer (included); and (4) photoreceptor layer (PRL), as the distance from the inner aspect of the retinal pigment epithelium (RPE) to the outer limiting membrane ( Figure 2 ). These measures were performed at the center of the fovea and at 1000 μm of eccentricity from the center along the OCT 9-mm central line in the nasal, temporal (horizontal line), and superior quadrant (vertical line). We computed as previously described by others the superior foveal-to-perifoveal ratio for the inner retinal layers (foveal-to-perifoveal IRL ratio) and for the photoreceptor layers (foveal-to-perifoveal PRL ratio) measurements. Briefly, the foveal-to-perifoveal ratios are computed by dividing the values of a studied layer thickness, here the inner and photoreceptor layer, at the foveal center and at 1000 μm of eccentricity along the vertical scan line in the superior quadrant. These ratios have been used to respectively describe the centrifugal displacement of inner retinal layers from the fovea and the retinal outer layer widening at the fovea during embryogenesis. Finally, we measured the foveal pit depth (FPD) as the distance between a 1-mm line tangential to the retinal surface and the ILM at the center of the fovea ( Figure 2 ). This parameter is another marker of the centrifugal displacement of the inner retinal layers that occurs during embryogenesis. When this phenomenon fails to occur, a shallow foveal pit in adult patients is present, as reflected by a decrease of foveal pit depth.
Statistical Analysis
Categorical variables were expressed as numbers (percentages) and comparisons were performed using the χ 2 test or the Fisher exact test when appropriate. For continuous variables, mean ± standard deviation (SD) was provided, and means were compared using the Mann-Whitney test. Correlations were performed using the Spearman correlation coefficient. P < .05 was considered significant. Analyses were performed using XLSTAT software (Addinsoft, Paris, France) version 2014.4.06.
Molecular Analysis
Molecular analysis of the VCAN gene have been conducted as previously described. In brief, DNAs were extracted from venous blood samples, using the QIAamp DNA mini kit (Qiagen, Valencia, California, USA). Intron-exon boundaries for exons 6, 7, 8, and 9 of the VCAN gene were amplified by polymerase chain reaction. Sanger sequencing reactions were performed in an automatic genetic analyzer (ABI PRISM 3100 genetic analyzer; Applied Biosystems, Foster City, California, USA) using the BigDye terminator cycle sequencing kit (DNA sequencing kit; Applied Biosystems).
Results
A total of 12 eyes of 9 patients from 3 unrelated families with a clinical picture of Wagner syndrome and carriers of a pathogenic VCAN mutation were included in this study; and 28 eyes from 15 unaffected family members, VCAN mutation negative, were selected to serve as controls (see Figure 1 for inclusion flow chart details).
The clinical and molecular data of all included patients with Wagner syndrome are summarized in Table 1 with corresponding references to our previous descriptions.
Baseline characteristics of both groups are shown in Table 2 . In summary, no significant differences were found for age (31 ± 18 3 years and 37 ± 22 6 years) in the control and Wagner groups ( P = .5; Mann-Whitney test) or for sex ratio (male-to-female ratio was, respectively, 8:7 and 4:5 [ P = .6; χ 2 test]). Spherical equivalent (−0.3 ± 1.2 diopters and −3.5 ± 2.8 diopters) as well as axial length (23.4 ± 0.7 mm and 24.8 ± 1.7 mm) were significantly different between the control group and Wagner group ( P =.001 and P = .026, respectively). BCVA was 0 ± 0 logMAR units for control patients and 0.2 ± 0.16 logMAR units for patients with Wagner syndrome ( P = .0001). Among the patients with Wagner syndrome, 6 were pseudophakic and 6 were phakic with a clear crystalline lens. We found no significant difference between phakic and pseudophakic patients regarding all the above studied parameters.
| Control Group | Wagner Group | P Value | |
|---|---|---|---|
| Number of patients | 15 | 9 | |
| Number of eyes | 28 | 12 | |
| Age (y, mean ± SD) | 31 (± 18.3) | 37.5 (± 22.6) | .5 NS |
| Male-to-female ratio | 8:7 | 4:5 | .67 NS |
| BCVA (logMAR ± SD) | 0 ± 0 | 0.2 ± 0.16 | .0001 |
| Spherical equivalent (diopters, mean ± SD) | −0.3 (± 1.2) | −3.5 (± 2.8) | .001 |
| Axial length (mm, mean ± SD) | 23.4 (± 0.7) | 24.8 (± 1.7) | .026 |
Qualitative SD OCT assessment showed in 10 eyes (10/12, 84%) from 8 patients with Wagner syndrome a macular thick hyperreflective structure attached to the perifovea in an annular aspect with no or limited attachment to the inner (superficial) layer of the underlying fovea, sometimes forming a bridge over the foveal pit. The outer (deep) aspect of the multilayered membrane was either partially detached ( Figure 3 ) from the retina (posterior vitreous detachment) or attached ( Figure 4 ). Total absence of this posterior membrane was observed for only 1 patient with Wagner syndrome (individual C.III.1), but abnormal vitreoretinal interface abnormalities in the form of vitreoschisis were present in the perifoveal region of both of her eyes ( Figure 4 , Bottom). Of note, 1 patient with Wagner syndrome (individual A.IV.7) had an optic nerve inverted nasally, or situs inversus of the optic disc and retinal vessels ( Figure 4 ).



