Retrochiasmal Visual Pathways and Higher Cortical Function
Jason J. S. Barton
Matthew Rizzo
CEREBRAL VISUAL LOSS: AN OVERVIEW OF CAUSES
The brain regions that process vision cover a broad portion of the posterior cerebral hemispheres, and cerebral lesions frequently produce visual disturbances. The most frequent cause is cerebrovascular disease, particularly embolic or thrombotic infarction in the posterior cerebral artery territory (Chapter 4, Fig. 15). Several other vascular mechanisms are possible but less common (Table 1). Transient ischemia may also play a role in the aura of classic migraine, though permanent migrainous visual deficits are rare.
TABLE 1. Causes of Cerebral Visual Loss | |
|---|---|
|
Trauma is another frequent cause of visual cortical lesions. Direct injury to visual areas occurs from open head injury with penetration of the skull from falls, falling objects, knives, bullets, or blows. Closed head injury may involve rapid acceleration and deceleration of the head, as in a fall or when the head strikes the dashboard in a car crash. The brain continues forward due to inertia, then moves back in the opposite direction. A coup injury produces bruising on the side of the impact and a contre-coup injury on the opposite side. Thus trauma to visual areas may follow a blow to either the occiput or the forehead.
Both cerebrovascular and traumatic brain injury can indirectly injure visual areas through cerebral edema. As the skull is a closed compartment, edema can result in herniation syndromes. Tentorial herniation of the uncus may compress the posterior cerebral arteries, infarcting one or both occipital lobes, with severe and permanent visual deficits in survivors.
There are many other causes of cerebral visual loss (Table 1). Vision impairment is frequent in Alzheimer’s disease, the most common cause of dementia in older adults, and can even be the presenting complaint in patients with the visual variant of this disorder. This group may perplex the eye specialist unaware of this manifestation of Alzheimer’s disease and who has few tools for measuring the relevant visuoperceptual and cognitive impairments. Creutzfeldt-Jakob disease, a rare, rapidly progressive, and incurable disorder, may likewise present with visual complaints prior to dementia, myoclonus, and the development of triphasic waves on the electroencephalogram (EEG). In these cases, there are concerns about transmitting the infectious agent to other patients through inadequate sterilization of instruments.
Finally, in addition to increased incidence of pathologic impairments of the eye and brain in the elderly, visual performance is also reduced through normal aging. Reduction in the speed of neural processing in the central nervous system results in cognitive slowing, reduced attention capacity, and shrinkage in the ‘useful field of view’ despite apparently normal visual fields on conventional perimetry. Such visual impairment can affect daily activities such as reading, route finding, face recognition, and driving, and may increase the risk of injury from car crashes and hip fractures.
LESIONS OF THE RETINO-GENICULO-STRIATE PATHWAY
Knowledge of the gross anatomic relations, vascular supply, and retinotopic and functional anatomy of the retino-geniculo-striate pathway is essential to understand the visual defects and associated signs from lesions of these structures. These aspects of vascular and neural anatomy are discussed in Chapter 4.
Optic Tracts
The optic tract is the continuation of the anterior visual system from the optic chiasm to the lateral geniculate nucleus (LGN). Only the contralateral hemifield is represented. The decussated nasal retinal fibers are not initially well aligned topographically with the other eye’s temporal retinal fibers, but retinal correspondence improves toward the termination of the tract at the LGN. The retinotopic map is also tilted in the optic tracts, so that the macula is represented dorsally, inferior retina (superior visual field) laterally, and superior retina (inferior visual field) medially1 (Chapter 4, Fig. 9). The magnocellular and parvocellular axons may also be segregated, with magnocellular axons more ventral.2 Both of these topographies are mirrored in the LGN. The main vascular supply of the optic tract is the anterior choroidal artery.
The functional anatomy of the optic tract is reflected in several important clinical findings. First, partial lesions cause contralateral homonymous defects that can be markedly incongruous (different patterns of visual loss in the two eyes), due to the poor topographic alignment of the two retinal maps (Figs. 1 and 2).3,4 In contrast, lesions of the optic radiations cause only mild incongruity, and striate lesions are highly congruous. Complete transection of the optic tract will lead to congruous complete hemianopia, though this is less frequent than partial lesions. Reduced acuity with an optic tract lesion indicates bilateral tract damage or extension of the lesion to the optic chiasm or optic nerves.4,5
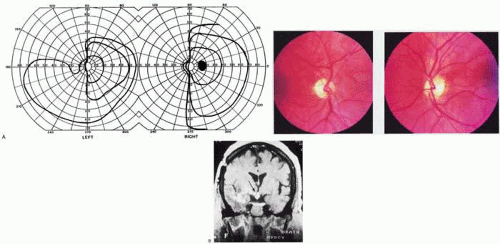 Fig. 1. Optic tract lesion. A 53-year-old woman with CNS sarcoidosis diagnosed 7 years previous with focal motor seizures. She had no visual symptoms. Acuity was 20/40 in both eyes. A: There was RAPD in the left eye, visual fields showed incongruous left hemianopia. B: Fundoscopy showed bow-tie atrophy in the left eye and temporal atrophy in the right eye. C: MRI showed enhancing lesions of right temporal lobe, infundibular region, and right optic tract. (From Rosen ES, Eustace P, Thompson HS, Cumming WJK [eds]: Neuro-Ophthalmology. London: Mosby, 1998.) |
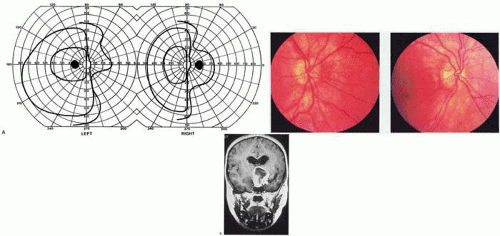 Fig. 2. Optic tract lesion. An 8-year-old girl with 1 month of headache and horizontal diplopia from right VI nerve palsy. Acuity was 20/25 in both eyes, and there was RAPD in the right eye. A: Visual fields showed mildly incongruent right hemianopia. B: She had twin-peaks papilledema in the right eye and nasal disc swelling in the left eye. C: MRI showed parasellar mass, involving left more than right, which was a hypothalamic glioma. (From Rosen ES, Eustace P, Thompson HS, Cumming WJK [eds]: Neuro-Ophthalmology. London: Mosby, 1998.) |
Second, because the axons in the optic tract originate from retinal ganglion cells, damage to the tract will cause optic atrophy over time. This is present in both eyes, but because only half or less of the axons of each eye are affected, the atrophy is less severe than with most optic neuropathy. Also, the pattern of optic atrophy will differ between the eyes. In the eye with temporal field loss, the axons from the nasal retina are affected. The fibers from the nasal periphery enter the nasal side of the disc, while those from the nasal macula enter the temporal disc in the papillomacular bundle. Atrophy is seen in these nasal and temporal wedges of the disc, but the superior and inferior sectors are spared because these contain fibers from the temporal retina. The result is “bow-tie” or “band” optic atrophy (Fig. 1). In contrast, atrophy in the eye with nasal field loss will affect the superior and inferior wedges and the papillomacular bundle, but not the nasal wedge. This will appear as diffuse or temporal disc pallor. These patterns of atrophy can also be demonstrated with optical coherence tomography.6 Another distinctive optic disc picture occurs in the eye with temporal field loss when a mass lesion causes papilledema and compresses the optic tract. Disc swelling occurs in the superior and temporal disc but not in the atrophic bow-tie regions, creating “twin-peaks” papilledema (Fig. 2).7,8
Third, because fibers for the pupillary reflex also travel in the optic tract, there is often a relative afferent pupil defect (RAPD). With a significantly incongruous hemianopia, the RAPD may be in the eye with greater visual loss. With a complete tract lesion the RAPD will be in the eye with temporal field loss4,9 because the temporal hemifield is larger and there are slightly more axons from the nasal than temporal retina (ratio 53:47). The RAPD is a useful sign in optic tract hemianopia as it may be present at a time when optic atrophy has not yet developed (Chapter 15).10
Other reported pupillary abnormalities include Wernicke’s hemianopic pupil, which is an intraocular afferent pupil defect, with less pupillary constriction from light on the hemianopic hemiretina compared to light on the intact hemiretina. Wernicke’s hemianopic pupil is difficult to elicit at the bedside because of intraocular light scatter,3 though it may be seen with computerized pupillometry (Fig. 3).
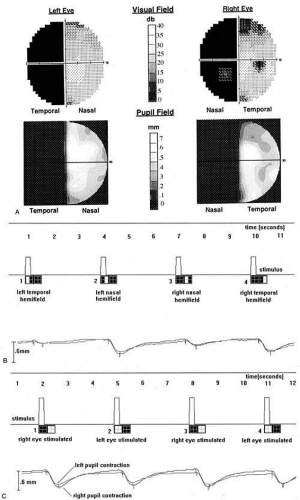 Fig. 3. Pupil perimetry results in optic tract lesions. A patient with a right optic tract lesion has incongruous left homonymous hemianopia (A, top) and lacks pupillary responses to focused light in the blind hemifield (A, bottom). This is also shown with hemifield light stimulation (B), where light in the left eye’s temporal hemifield (pulse 1) and the right eye’s nasal hemifield (pulse 3) elicits much less pupilloconstriction than light in the seeing hemifields (pulses 2 and 4). In addition, pupillometry with full-field stimuli (C) shows smaller amplitudes of pupil constriction to light in the left eye (pulses 2 and 4) than in the right eye (pulses 1 and 3), confirming a left RAPD. (Courtesy of Dr R Kardon, University of Iowa.) |
The combination of optic atrophy, RAPD, and field incongruity is important to recognize with homonymous hemifield defects because it changes the differential diagnosis of hemianopia (see Fig. 14). Most hemifield defects from lesions of striate cortex or the optic radiations are due to vascular disease or other intracerebral pathology. Most optic tract lesions are compressive extrinsic masses, with a differential diagnosis similar to that for optic chiasmal lesions (Chapter 6). In fact, patients with combined damage to the optic tracts, chiasm, and nerve are not rare.4,11,12,13,14 Pituitary adenomas, giant aneurysms of the internal carotid artery, meningiomas, and craniopharyngiomas are the chief causes of optic tract dysfunction. The investigation of choice is imaging of the parasellar region, with coronal and axial sections and contrast administration.
Less common lesions include inflammatory conditions such as multiple sclerosis3,12,15,16 and sarcoidosis (Fig. 1).17 Intrinsic optic pathway gliomas may occur in the optic tracts. Vascular lesions are rare, but there are reports of cavernous angiomata11,18 or arteriovenous malformations.19 Optic tract infarction can complicate anterior temporal lobectomy, possibly from vasospasm of the anterior choroidal artery.20 Trauma can affect the optic tract.3 Radiotherapy of pituitary tumors may be followed years later by optic tract necrosis.14 Optic tract dysfunction is a side effect of alpha-interferon.21 On occasion there is congenital absence of the optic tract22; such patients are often unaware of their hemianopia.
Lateral Geniculate Nucleus
The LGN is a subnucleus in the ventroposterolateral corner of the thalamus. Neighboring thalamic subnuclei include the medial geniculate nucleus ventromedially, the ventral posterior nucleus dorsomedially, and the pulvinar superiorly and dorsally. The medial geniculate nucleus gives rise to the acoustic radiations, which pass by the dorsomedial aspect of the LGN on their way to the auditory cortex in the temporal lobe. The optic radiations arise from the dorsolateral surface of the LGN. Ventrally, the hippocampus and parahippocampal gyrus face the LGN across the ambient cistern and the inferior horn of the lateral ventricle. The LGN has a dual blood supply: the anterior choroidal artery, a branch of the internal carotid artery, and the lateral choroidal artery, a branch of the posterior cerebral artery. The anatomy of the vascular territories within the LGN has been debated. Initial studies suggested that the anterior choroidal artery supplied the medial LGN as well as the optic tract and the lateral choroidal artery the lateral LGN. However, experience with surgical arterial lesions concluded that the anterior choroidal artery supplied both the lateral and medial aspects and the lateral choroidal artery supplied the hilus and mid-zone of the LGN.
In addition to its function as a relay in the visual pathway, the LGN is also a site of modulation, by back-projections from visual cortex24,25 and afferent projections from the brainstem reticular formation and superior colliculus.26 Some of the corticofugal input influences the stimulus selectivity of LGN neurons.25 Others postulate that these nonretinal inputs play a role in gating visual transmission through the LGN and thus participate in selective attention.24
The LGN is a triangular-shaped structure with six roughly horizontal layers containing segregated inputs from the two eyes (Chapter 4, Fig. 8). The ventral two layers are the magnocellular layers, whereas the other four layers are the parvocellular component; these differ in many structural and functional aspects (Chapter 4). The LGN has a retinotopic pattern, which is a continuation of that found in the optic tract. The macula is represented in a dorsal wedge including the hilum and projecting posteriorly, whereas the most peripheral fibers are located ventrally. Superior retinal fibers (contralateral inferior visual quadrant) are in the medial horn, and inferior retinal fibers (contralateral superior visual quadrant) are in the lateral horn.
Because the LGN is small and relatively secluded, lesions here are rare. Its intimate relation to the optic tract and optic radiation makes it difficult to be certain that a visual defect is due to LGN damage rather than damage to these structures. Indeed, visual field defects from purported LGN lesions resemble visual field defects from optic tract or optic radiation lesions.
Three main types of hemianopic defects have been described. The first is an incongruous hemianopia, much like that seen with optic tract lesions, reflecting the continued segregation of ocular inputs in the LGN. The other two patterns are sectorial hemianopias, reflecting the unusual territorial division between the anterior and lateral choroidal arterial supplies. With lateral choroidal ischemia, the hilum and middle zone of the LGN are affected, causing a wedge-shaped visual defect straddling the horizontal meridian (Fig. 4). 27 With anterior choroidal ischemia, the lateral and medial tips of the LGN are infarcted, resulting in the reverse defect, loss of the superior and inferior aspects of the contralateral hemifield with sparing around the horizontal meridian.28,29 Unusual cases of presumed bilateral LGN damage have presented with an “hourglass” shape to either the visual field defect30,31 or the region of spared vision.32
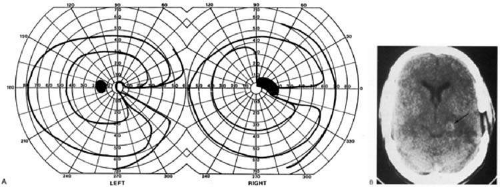 Fig. 4. Lesion of lateral geniculate nucleus. Slightly incongruent right sectoranopia along the horizontal meridian, with hemorrhage in the vicinity of the LGN. This field defect would result from damage to the mid-zone of the LGN. (From Rosen ES, Eustace P, Thompson HS, Cumming WJK [eds]: Neuro-Ophthalmology. London: Mosby, 1998.) |
Optic atrophy often accompanies LGN lesions. If there is damage to almost all the LGN, the optic atrophy will have a similar appearance to that seen with optic tract lesions. If there is partial damage causing sectorial hemianopias, then the optic atrophy may be more subtle and restricted to the relevant sectors of the disc.27,28 However, because the afferent fibers subserving the pupillary light reflex have already departed for the pretectum, there is no RAPD with lesions of the LGN. With incongruous hemianopia and optic atrophy, this is the only feature that permits distinction between optic tract and LGN lesions.
A variety of pathologies have been reported with LGN lesions. Infarction is the most likely cause of sectoral hemianopia, given the dependence of such defects upon the vascular anatomy,27,28 but astrocytomas and arteriovenous malformations are also reported. The LGN also appears to be a target of central pontine myelinolysis, a syndrome associated with excessively rapid correction of hyponatremia.30,31,33 LGN damage is even rarely a parainfectious complication of traveler’s diarrhea.32
Optic Radiations
The optic radiation may be affected anywhere in its course (Chapter 4); the type of visual field defect reflects the site of damage. Ischemic or hemorrhagic lesions of the internal capsule affect the optic radiation while it is still a relatively compact bundle, usually causing a complete homonymous hemianopia. A similar defect can arise from damage close to the termination in striate cortex (Fig. 5). Lesions of the ventral fibers in the anterior temporal lobe (Meyer’s loop) cause a contralateral superior visual quadrant defect (Fig. 6). Most often this defect aligns on the vertical meridian, with variable extension toward the horizontal meridian and central vision.34,35 Modern estimates from anatomic dissections and field defects after temporal lobectomy suggest that the most anterior extent of the Meyer’s loop is about 2.5 cm posterior to the tip of the temporal lobe.35,36,37,38,39 Lesions of the dorsal fibers in the parietal lobe cause an inferior visual quadrant defect (Fig. 7). Because there is no sharp demarcation of the dorsal fibers from the ventral fibers in this portion of the posterior pathway, the defect seldom aligns along the horizontal meridian.34 Overall, quadrantanopia is more frequent with lesions of striate cortex.34 Lesions of the temporal lobe more than 7–8 cm posterior to its anterior tip can affect both upper and lower radiations.35 Small lesions may also affect certain portions of the radiations and spare others: for example, damage to the midportion of the optic radiation can mimic the sectoranopias of LGN lesions40 (Fig. 8). Although there can be some incongruity to the visual field defects of optic radiation lesions, with about 15% greater field loss in the eye with the nasal hemifield defect,35 this is less marked than the incongruity with optic tract lesions.
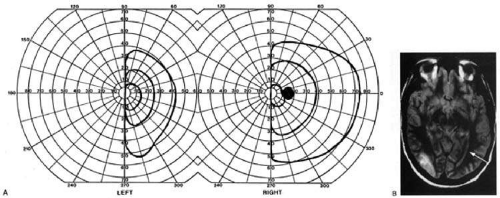 Fig. 5. Macula-splitting hemianopia. A 47year-old man with AIDS and sudden onset of poor vision. A: Fields show complete left hemianopia. B: MRI shows lesion of right lateral occipital cortex, affecting distal optic radiations. Biopsy showed nonspecific encephalitis. |
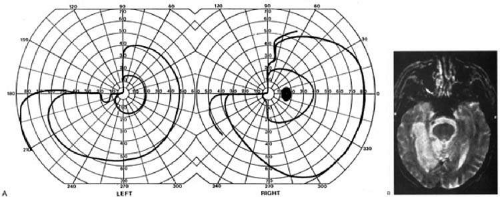 Fig. 6. Lesion of temporal optic radiation. A: Left superior quadrantanopia, respecting horizontal meridian, from infarct of right medial temporal lobe, (B) in posterior cerebral artery territory. (From Rosen ES, Eustace P, Thompson HS, Cumming WJK [eds]: Neuro-Ophthalmology. London: Mosby, 1998.) |
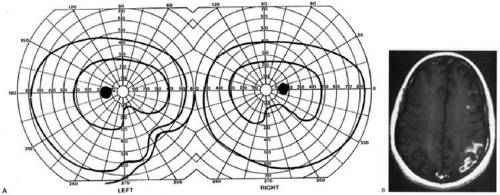 Fig. 7. Lesion of parietal optic radiation. A 35-year-old woman with 3 weeks of left-sided headache. A: Visual fields showed partial right inferior quadrantic defect, (B) MRI showed infarct of parietal lobe, involving optic radiations. (From Rosen ES, Eustace P, Thompson HS, Cumming WJK [eds]: Neuro-Ophthalmology. London: Mosby 1998.) |
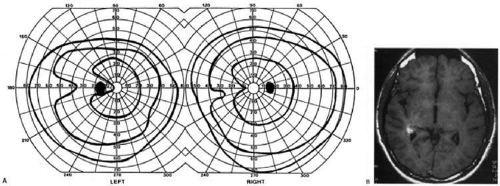 Fig. 8. Optic radiation sectoranopia. A 25-year-old man with prior history of intracerebral hemorrhage. A: While he had no visual symptoms, fields show subtle left sectoranopia straddling the horizontal meridian. B: MRI shows periventricular hemorrhage from a cavernous angioma, affecting the mid-portion of the optic radiations. |
Unlike lesions of the retino-geniculate pathway or the LGN, lesions of the geniculostriate axons do not lead to optic atrophy (with the exception of some congenital lesions, through trans-synaptic degeneration, as confirmed recently with optical coherence tomography)41 or pupillary defects. However, there are frequently other signs of cerebral damage,34 especially if the lesion is large. Thus, temporal lobe lesions cause superior quadrantic defects and sometimes also complex partial seizures, auditory or complex visual hallucinations (some of which may be seizures also), memory problems, or a Wernicke’s aphasia if the dominant hemisphere is involved. Parietal lesions with mainly inferior quadrantic defects may cause cortical sensory disturbances, such as impaired two-point discrimination and graphesthesia and impaired smooth pursuit toward the side of the lesion. With dominant hemisphere lesions, Gerstmann’s syndrome (acalculia, finger anomia, right-left disorientation, and agraphia) may occur, as can a variety of aphasic syndromes, including alexia with or without agraphia, Wernicke’s aphasia, or global aphasia.
The differential diagnosis of optic radiation lesions reflects the variety of cerebral hemispheric pathologies. Unlike lesions of the optic tract, most are infarcts in the posterior cerebral or middle cerebral artery territories. Tumors, vascular malformations, infections, and leukodystrophies are also possibilities. The temporal profile of the illness will often be the major clue to the etiology. The optic radiations are also often involved by surgical procedures. Because of this, there has been recent interest in using a new magnetic resonance imaging (MRI) technique, diffusion-tensor imaging, to map the optic radiations as part of preoperative planning,42,43,44 although there is some concern over whether its estimate of the most anterior portion of the Meyer’s loop is too conservative.45 Nevertheless, diffusion-tensor imaging may predict field defects after lobectomy.46
Striate Cortex
The primary visual area in the medial occipital lobe goes by several names: Brodmann’s area 17, visual area 1 (V1), calcarine cortex, and striate cortex (Chapter 4). The exact position of striate cortex varies between individuals. Although the parieto-occipital fissure forms a reasonably reliable anterior dorsal boundary, the posterior limit containing the macular representation is more variable, extending from the medial occipital surface over the first 1 to 2 cm of the posterior surface of the occipital lobe (Chapter 4, Fig. 10).
The main vascular supply of striate cortex derives from the posterior cerebral artery (Chapter 4, Fig. 15). A parieto-occipital branch supplies the superior calcarine bank, a posterior temporal branch supplies its inferior bank, and a calcarine branch supplies the central region posteriorly. Individual variation exists, however.47 Perhaps most importantly, the occipital pole is at the junction (watershed zone) of the vascular territories of the posterior and middle cerebral arteries, and there is again marked variation as to which artery supplies the foveal representation in striate cortex.47
The retinotopic arrangement in striate cortex is well known (Chapter 4) and confirmed with recent imaging studies of lesions.48 The foveal representation is posterior, at the occipital pole, and the far peripheral field is anterior, on the medial occipital surface.49,50 The superior bank of the calcarine fissure receives input from the inferior visual field, while the inferior bank contains the representation of the superior visual field. The most anterior part of striate cortex represents the monocular temporal crescent, the region of temporal field in the contralateral eye that lies beyond the limits of the nasal field (60°) of the ipsilateral eye. As in most of the visual system, there are fewer neurons devoted to peripheral vision than to central vision: over half of striate cortex is devoted to the central 10° (cortical magnification).48,51 Occipital cortex contains a mixture of monocular and binocular cells arranged in ocular dominance columns, but large separations between the inputs of the two eyes are not present.
Visual field defects from striate lesions.
Focal destruction of striate cortex produces a homonymous contralateral visual hemifield defect. Unlike the scotomata from lesions of the optic radiations and especially the optic tracts, the hemianopic defects from striate lesions are highly congruent, with virtually identical defects in the two eyes.
Complete destruction of striate cortex causes complete visual loss in the contralateral visual hemifield. Because this involves not only peripheral vision but also the contralateral half of the foveal region, it is called a macula-splitting homonymous hemianopia. This may occur with posterior cerebral artery ischemia in a patient whose entire striate cortex is supplied by that artery. Macula-splitting hemianopias can occur with complete lesions anywhere along the retrochiasmal visual pathways, and thus lack localizing value (Fig. 5). Other signs may help in localization. Reading is particularly impaired by involvement of the central 5°.52
Partial lesions of the striate cortex are frequent. With posterior cerebral infarcts, a macula-sparing hemianopia occurs in patients with adequate collateral circulation of the macula region (occipital pole) from the middle cerebral artery47 (Fig. 9). Previously, macula sparing was thought to be due to bilateral representation of a small stripe flanking the vertical meridian, which expanded to as much as 3° at the fovea.53 However, studies of monkey V1 do not find bilateral representation of the hemimaculae,54 and computed tomography (CT) and MRI studies in humans with hemianopia document the correlation of macular sparing with sparing of the occipital pole.55,56 Also, careful perimetry of hemianopes with the scanning laser ophthalmoscope shows that, while there is a slight overlap from the seeing field into the blind field along the meridian, macular sparing of 2° to 5° is only present in some patients.57,58 Therefore, sparing more likely reflects the extent of occipital pathology than retinal anatomy. Macula sparing has some localizing value, in that it is rarely found with lesions outside of striate cortex. Hence, its presence indicates a high probability of an occipital lesion.
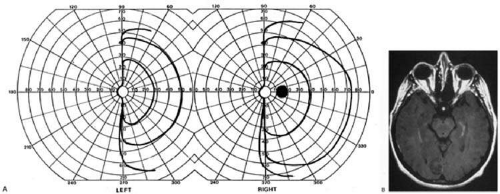 Fig. 9. Macular-sparing striate hemianopia. A 49-year-old woman with headache and difficulty seeing to the left for 1 week. A: Fields show left hemianopia that spares a small zone around the central fixation spot. B: MRI shows infarct of right striate cortex, with sparing of occipital pole. (From Rosen ES, Eustace P, Thompson HS, Cumming WJK [eds]: Neuro-Ophthalmology. London: Mosby, 1998.) |
The upper and lower banks can also be involved separately. Ischemia can do this because the banks have separate blood supplies. Upper bank infarcts cause homonymous contralateral inferior quadrantanopia (Fig. 10), and lower bank infarcts cause superior quadrantanopia. Although altitudinal defects have been reported occasionally,59,60 most quadrantic defects do not align at the horizontal meridian because the upper field merges without interruption into the lower field in the depths of the calcarine fissure. Thus, it has been argued that quadrantic defects that respect the horizontal meridian are due to involvement of area V2, surrounding striate cortex61; this remains controversial. Quadrantanopsias are three times more common with striate lesions than with optic radiation lesions.34 Striate quadrantanopsias are more frequently isolated deficits but can be associated with other signs of higher cortical visual dysfunction, such as pure alexia or hemiachromatopsia; whereas optic radiation quadrantanopsias are usually accompanied by hemiparesis, dysphasia, or amnestic problems.34
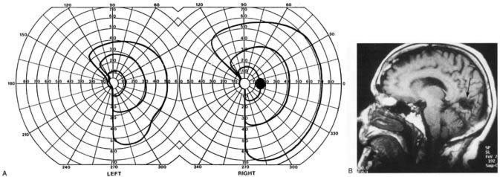 Fig. 10. Striate quadrantanopia. A: A 68-year-old woman with a stroke 3 years previous, causing left inferior quadrantanopia. B: MRI shows infarct of the superior bank of the right calcarine cortex. |
Selective lesions can also occur along the anterior-posterior extent of striate cortex. A lesion of the occipital pole alone causes homonymous central hemiscotomata (Fig. 11).56,62 This can occur with watershed infarcts during systemic hypoperfusion. Slightly more anterior lesions in the middle zone of striate cortex cause homonymous peripheral scotomata (Fig. 12). The highly congruent, homonymous nature of these defects and their restriction to one hemifield differentiate these from ocular causes of central or paracentral visual loss. Lesions with such small field defects can be missed on CT.55 MRI with coronal sections through the occipital lobes should be performed, although even this may miss small lesions, particularly at the occipital pole.
 Fig. 11. Central hemiscotomata. A 37-year-old man with sudden onset of difficulty reading. A: Fields show small inferior central homonymous scotomata restricted to right hemifield, respecting the vertical meridian. B: MRI shows subtle hypointensity at occipital pole, presumably a small infarction. (From Rosen ES, Eustace P, Thompson HS, Cumming WJK [eds]: Neuro-Ophthalmology. London: Mosby, 1998.) |
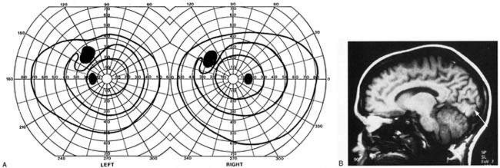 Fig. 12. Homonymous peripheral scotomata. A 32-year-old woman with classic migraine history. Two weeks prior she had a severe migraine with transient left-sided weakness and typical blurring in left hemifield, which on this occasion did not resolve. A: Fields show small homonymous scotomata in left upper quadrant. B: MRI shows small striate infarct of the midzone of the inferior bank of the calcarine fissure. |
A near-complete lesion that spares only the most anterior portion of V1 causes an interesting field defect, hemianopia with sparing of the monocular temporal crescent (Fig. 13). The hemianopia involves the whole nasal hemifield of the ipsilateral eye, but the temporal hemianopia of the contralateral eye spares a crescent-shaped island of vision in the far periphery.63 This is the monocular temporal crescent, the region of the visual field that is represented in the temporal field of one eye but not the nasal field of the other. The initial sense of incongruity may raise suspicions of an optic tract lesion; however, the absence of optic atrophy and RAPD, the high congruity of the homonymous defect inside 60°, and the location of the crescent outside 60° eccentricity indicate that the lesion is likely in striate cortex. The converse defect, a monocular temporal crescentic scotoma, can occur with a lesion of cortex just behind the splenium of the corpus callosum, along the parieto-occipital sulcus.64
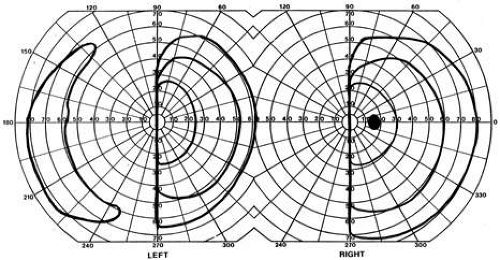 Fig. 13. Sparing of the monocular temporal crescent. A 65-year-old man with left hemianopia from right medial occipital infarct. Fields show complete hemianopia within an eccentricity of 60°, but sparing of the temporal field beyond 60°, which is normally only perceived in the temporal field. This indicates a striate lesion with sparing of the retrosplenial portion of calcarine cortex. (From Barton JJS, Sharpe JA: Smooth pursuit and saccades to moving targets in blind hemifields. Brain 120:681, 1997.) |
Most striate lesions are infarction, mainly from posterior cerebral artery occlusion (Fig. 14), with sudden onset visual loss and often headache.65 In about half of all patients, the visual field defect is the only deficit,65 but in others damage to medial occipito-temporal regions causes amnesia, prosopagnosia, and color perception defects. A syndrome of agitated delirium and hemianopia occurs with lesions of the medial occipital lobe, parahippocampus, and hippocampus.66,67,68 Brainstem signs include impaired level of consciousness, III nerve palsy, dysarthria, and hemiplegia.65 Causes of ischemia are most frequently cardiac emboli and vertebrobasilar occlusive disease; migraine is a rare cause of permanent defects.65 Hemorrhage, vascular malformations, and primary and secondary malignancies are much less common.34
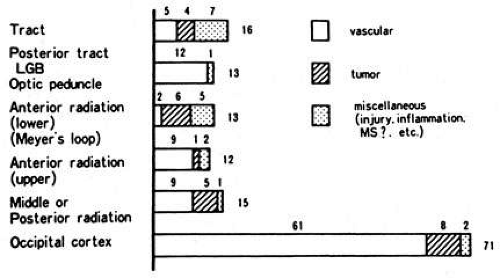 Fig. 14. Location and etiology of homonymous hemianopia in 140 patients. (From Fujino T, Kigazawa K, Yamada R: Homonymous hemianopia. A retrospective study of 140 cases. J Neuroophthalmol 6:17, 1986, Aeolus Press, with permission.) |
Bilateral lesions of striate cortex are not rare. Focal midline lesions such as tumors or traumatic injury may affect both striate cortices concurrently, since the right and left striate cortices face each other on the medial occipital surface. The most common cause, however, is posterior circulation ischemia.69 This can affect both striate cortices either simultaneously or sequentially,69 as the right and left posterior cerebral arteries have a common origin from the basilar artery. Twenty-two percent of patients with a unilateral occipital infarction will develop bilateral infarction over 3 years.70 Bilateral incomplete hemianopia is distinguished from bilateral optic nerve or ocular disease by the high congruity of the visual fields and step defects along the vertical meridian that indicate the hemifield nature of the visual loss69 (Fig. 15). This is why it is important to seek a skilled perimetrist, but even then it can be difficult to demonstrate with bilateral hemiscotomata from occipital pole lesions.71 Bilateral quadrantanopsias can occur,59,60 often in patients with prosopagnosia and achromatopsia, for example, and may mimic the altitudinal defects of optic neuropathy.
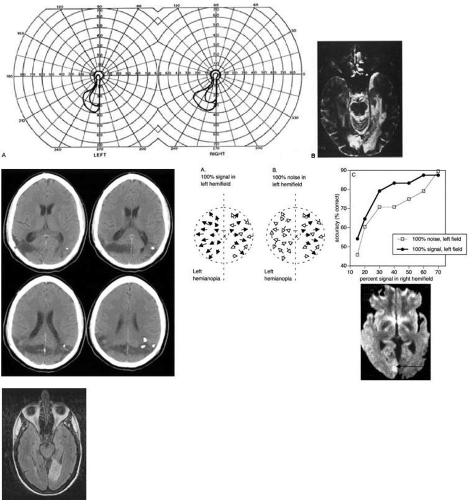 Fig. 15. Bilateral incomplete hemianopia. A 69-year-old man with decreased vision after prostate surgery. A: Fields show remaining central parafoveal vision with some sparing of inferior left quadrant. Note how the defect respects the vertical meridian. B: MRI shows bilateral medial occipital infarction, with sparing of the occipital poles, accounting for the macular-sparing bilaterally. (Courtesy of Dr Lucia Vaina.) C: CT scan of man with cerebral blindness after a gunshot wound. D: Interactions between blind and seeing hemifields. Bottom shows the diffusion weighted MRI of the small right striate lesion, causing left hemianopia. Left shows the two contrasting stimuli. The patient’s task is to judge the direction of the optic flow motion (black arrows) in the seeing right hemifield, from amid a background of moving noise (white arrows). In the blind field there is either random noise or a strong optic flow motion pattern. Results on right show that the accuracy of the patient at all levels of mixed noise and optic flow are improved by 5% to 10% when there is a strong flow pattern in the blind field. E: MRI scan of patient with stroke causing a right hemiachromatopsia as well as partial superior quadrantanopia. |
Cerebral blindness.
Cortical blindness is a loosely used term, at times referring to visual loss from occipital lobe damage, even if the loss is incomplete. Hence, hemianopia or bilateral quadrantanopia has been called cortical blindness. It is best reserved for bilateral complete or severe hemianopia, with acuity at light perception only or worse, and no detectable peripheral vision. Since lesions may involve both gray and white matter, a better term is cerebral blindness.
Cerebral blindness can be persistent or transient. The most frequent cause of persistent cerebral blindness is cerebrovascular infarction.72 In addition to the common causes of emboli or atherosclerosis, it has also been reported with vertebrobasilar arteritis,73 subclavian steal,74,75 and hypotension from antihypertensive medication.76,77 Cerebral blindness can complicate cardiac surgery, through hypotension or emboli.72 A rare vascular cause is rupture of occipital mycotic aneurysms with endocarditis.78
Cerebral blindness is distinguished from ocular disease by both normal pupillary light responses and normal funduscopic examination. These may lead to an erroneous diagnosis of factitious visual loss. Associated signs of damage to parietal or temporal structures help to confirm cerebral blindness but may not always be present. Visual evoked potentials are of limited diagnostic value. They can be altered voluntarily by subjects without visual loss79 and can be normal in patients with striate lesions.80,81 They cannot differentiate between blind and seeing children with neurologic disease,82 and normal or abnormal results do not predict visual outcome.72,83 Absent evoked responses are rare and may only occur early in the course.82 Absent alpha rhythm on EEG84,85 is reportedly a more sensitive diagnostic sign than abnormal visual evoked potentials.72 CT scans can be normal, but modern MRI with coronal images through the occipital lobe should reveal most striate or optic radiation lesions with complete and persistent visual loss (Fig. 15C). Single-photon emission computerized tomography (…(SPECT) scans may reveal bilateral functional defects in cases with unilateral MRI lesions.86
Among adults with infarction, blindness is permanent in 25% of patients,69 and residual visual field defects are common in the rest. Bioccipital CT lucencies carry a poor prognosis for recovery, but abnormal visual-evoked potentials do not correlate with severity or outcome.72 Although the abnormalities on visual-evoked potentials are not diagnostic, they tend to improve as vision returns.87,88,89
Transient cerebral blindness can last hours to days, often with full recovery (Table 2). Both ictal and postictal cerebral blindness are reported in children and adults.90,91,92 Transient cerebral blindness can occur with metabolic insults,93,94,95 hypertensive encephalopathy,96 hydrocephalus,97 trauma,84,98 and cortical venous thrombosis.99 Toxins are an important cause, especially chemotherapeutic agents.100,101,102,103,104 Cerebral blindness is associated with the iodinated contrast agents used in angiography;72,105,106 CT scans with contrast show disruption of the blood-brain barrier in the occipital lobes105,106 as early as one hour after angiography.107
TABLE 2. Causes of Transient Cerebral Blindness | |
|---|---|
|
The pathogenesis of transient cerebral blindness varies with the cause. Vasogenic edema is probably the chief factor in the posterior reversible encephalopathy syndrome (PRES) that is associated with seizures, chemotherapy, and hypertensive encephalopathy.108 Angiographic contrast agents may cause breakdown of the blood-brain barrier under osmolar stress and subsequent neurotoxic effects,107 providing a rationale for dexamethasone and mannitol as specific therapy in this context. MRI using diffusion-weighted imaging and apparent diffusion coefficient images can distinguish vasospasm from stroke and help determine the prognosis for recovery of visual loss.108
As well as vasogenic edema, vasospasm may play a role in head trauma,109 eclampsia,110,111 methamphetamine abuse,112 and meningitis, which may also induce vasculitis.89,113
Pediatric cerebral blindness is not uncommon. Children may not complain of visual loss but present with agitation, disorientation, and unsteadiness.85,109,114 During tests of vision they may not respond or may confabulate answers.114,115 Further observation shows that they do not fix on, follow, or make saccades to objects, blink to threat, or show optokinetic responses.82,85,115 There can be an associated strabismus. Major causes include head trauma, bacterial meningitis,88,89,113,115 and hypoxia from cardiac or respiratory arrest.115 Other causes include encephalitis, metabolic,95,115,116 and toxic112 conditions (Table 3). Seizures are common, and mental retardation is a frequent complication among patients with meningitis.88
TABLE 3. Causes of Pediatric Cerebral Blindness | |
|---|---|
|
Posttraumatic transient cerebral blindness is a syndrome that tends to occur in children and young adults.85,117,118 It occurs in about 1% of closed head injuries.84,98,119 The impact is often in the parieto-occipital region.84,85,118,120 Headache, confusion, anxiety, nausea, and vomiting are frequent,118 as is loss of consciousness at the time of injury.98 Blindness may occur immediately or after a few minutes,85,98,118,120 sometimes after a few hours in adolescents.98 Vision recovers within 24 hours,118,120 though the course in adults is more variable, with occasional residual deficits.98 CT of the head often is normal,114,118,120 but EEG shows bilateral occipital slowing.85 Circumstantial evidence for vasospasm in traumatic cerebral blindness is an association with prior migraine.98 Protracted recovery, especially in children, may indicate cerebral contusions120 or occipital lobe infarction from tentorial herniation.121 There can be a personal or family history of seizures and migraine.98
The course of cerebral blindness is variable. It can last hours, days, or weeks, patients recover fully, evolve into a hemianopia or other field defects, or blindness can be permanent.83,88,115 If there is recovery, it usually occurs within 5 months.83 Recovery of useful vision is more likely in children than adults.115 The prognosis is better with hypotension during cardiac surgery than with other causes.83 The prognosis with hypoxia is worst for preterm infants, who tend to have periventricular leukomalacia rather than parasagittal watershed infarctions. Optic radiation damage on their CT or MRI is a poor sign.122 Normal MRI in most other settings is a good prognostic sign,83,122 and significant recovery can occur if hypoxic damage is limited to the visual cortex.122 Spike-and-wave discharges on EEG are associated with multiple handicaps and poor visual recovery.83
Transient cerebral blindness occurs in about 1% to 15% of patients with pre-eclampsia or eclampsia.110,111,123 Fluctuations in blood pressure with peripartum blood loss may be a contributing factor.124 The pathogenesis is analogous to that of hypertensive encephalopathy, with development of petechial hemorrhages and focal edema from impaired cerebral vascular autoregulation and ischemia from vasospasm.111,125 CT scans may be normal or show bioccipital lucencies.111,126 MRI is more sensitive, showing T2 hyperintensities and T1 hypointensities similar to those in hypertensive encephalopathy.125,127 Management is that of eclampsia, with magnesium sulfate, fluid restriction, and blood pressure control. The prognosis is good, with virtually all women regaining vision within a few hours to a week, rarely as long as 3 weeks.111
The differential diagnosis of eclamptic visual loss includes cerebral venous thrombosis128 and systemic hypoperfusion due to pulmonary emboli.129 Antiphospholipid antibodies may be associated with infarction or venous thrombosis.130 Eclampsia can also affect the eyes, causing retinopathy with retinal detachment, edema, and vascular thrombosis,131 or, more rarely, anterior ischemic optic neuropathy.132 Pituitary apoplexy can present with severe headache and visual loss and is a medical emergency.
Anton’s syndrome.
About 10% of patients with cerebral blindness are not aware of their deficit and insist that they can see.69,72 This denial syndrome may be considered a form of anosognosia (denial of acquired impairment), which has been attributed to right hemispheric dysfunction or disconnections between the thalamus and the right parietal lobe.133 Denial of blindness can also occur in patients with ocular or optic nerve disease when there is concurrent dementia or a confusional state. In contrast, altered mental status is not a necessary accompaniment of denial of blindness with lesions of the visual cortex. Thus, Anton’s syndrome should be more specifically defined as the denial of blindness in the absence of dementia or delirium. However, even cases meeting this more restrictive definition do not always have a lesion of visual cortex, since such a presentation can also result from unusual combinations of bilateral optic neuropathy and bilateral frontal lobe disease.134
Abnormalities in the remaining visual field.
Some patients with hemifield visual defects complain of difficulties with their remaining vision, such as visual fatigue or blurring, especially in tasks with high attentional demands like reading, finding a face in a crowd, or driving a motor vehicle. There is evidence that unilateral lesions produce not only contralateral scotomata but also deficits in both the remaining ipsilateral and contralateral visual fields. There are spatial and temporal contrast sensitivity deficits in the supposedly normal ipsilateral hemifields of patients with homonymous hemianopia.135 There is reduced sensitivity and increased response times to signals in both hemifields and reduction in the “useful field of view.”136 Saccadic response times to visual targets in the ipsilateral hemifield are also reduced.137
The origins of these subtle bilateral effects of unilateral lesions are unclear. One hypothesis is that the inevitably associated white matter damage disrupts visual connections, including projections from V1 to other visual areas, callosal connections between the same or different visual areas in the two hemispheres, and feedback projections from a higher visual area to a lower one. A lesion in one area can thus affect the functioning of another, more distant area (diaschisis). Disrupted connections can impair the synthesis of information from other vision areas and both hemispheres, producing long-range disturbances in both hemifields, outside the limited contralateral scotoma. Damage to extrastriate regions may have similar bilateral long-range effects, as shown in monkeys with lesions of area V4, for example.137a Thus, associated extrastriate damage may contribute to impaired processing efficiency in the ipsilateral field of patients with hemianopia.136
Standard kinetic and automated static perimetry are not designed to detect such deficits. These tests ignore response speed and minimize the role of attention to get maximal estimates of sensory function, an effective approach for gauging the classic field defects due to dysfunction of the retinogeniculostriate pathway. However, the working visual capacity in elderly or brain-damaged individuals is better approximated by tests of vision under conditions of increased attentional load.136,138 Measures of this useful field of view (UFOV) have been designed, using central or peripheral distractors during perimetric tests of localization ability to gauge the effects of attention. UFOV evaluation may reveal defects associated not only with hemianopia but also with aging and Alzheimer’s dementia, where they may have practical import, in that UFOV reduction predicts crashes in driving simulations.139
Rehabilitation of homonymous defects.
Partial recovery within 2 to 3 months occurs in 15% of patients with hemifield defects.140 A scotoma that persists beyond several months is permanent. The larger and closer to fixation the scotoma, the worse the effects on daily activities such as reading and driving.141,142 Attempts at restoring the visual fields through visual exercises have been made in adults143,144,145,146 and children.147 The validity of some of these results has been questioned, as well as their interpretation, as to whether there was true recovery using neural plasticity, or shifts in the response bias of patients or their fixation.
It is possible to train individuals to scan the hemianopic field with eye movements,148 a strategic adaptation that also occurs without training as long as subjects do not have additional hemineglect.149,150 Adaptive changes to improve reading can also be learned.151 There are even some claims that these reading and scanning adaptations improve visual fields slightly,144,151 which need verification. Another potential adaptive treatment for the navigational behavior of hemianopes is the use of prisms to shift elements of the blind side of space into the seeing hemifield.152 While this distorts the field of vision to some degree, the ability to detect stimuli rapidly on the blind side can offer some advantages while walking.
Blindsight and residual vision.
Patients with visual loss due to lesions of the striate cortex or optic radiations may have some remnant visual function in their blind field. Some of these patients retain some awareness of visual stimuli and hence have “residual vision,”153,154,155 implying a severe relative field defect. Others deny awareness of stimuli, even though they perform better than chance when asked to indicate or guess at some property of the stimulus; these are said to possess “blindsight.”156,157,158 Whether the pathophysiology of residual vision differs from blindsight is unclear. The phenomenological distinction between the two is not necessarily sharp. Perceptual awareness varies along a spectrum: patients may retain a vague awareness of the presence of a stimulus but not of its particular features, which they can nonetheless discriminate or act upon.159 Stimulus parameters can be manipulated so that a patient shows residual vision under some conditions and blindsight under others.160,161
VARIETIES OF BLINDSIGHT PHENOMENA.
Some studies find that saccades to locations within hemianopic fields are weakly correlated with target position, mainly for a limited range of paracentral locations.156,162,163,164 Other studies have failed to find saccadic localization.137,165,166 Reaching and pointing are also usually weak and variable, sometimes only in patients with residual vision,155,167,168 but there are other reports of nearly normal manual localization.159,164
Target localization was easier with moving rather than stationary targets in some reports169,170 but not another.171 This is reminiscent of Riddoch’s phenomenon, in which movement is appreciated before static targets in recovering hemianopic defects.172 Studies of blindsight motion perception show some residual perception of the speed and direction of rapid bright spots.153,155,173 With larger stimuli such as optokinetic gratings, some patients can discriminate motion direction174 and a few may experience an illusion of self-motion.175 Some of these motion abilities may actually be secondarily derived from spatial position or flicker rather than true motion velocity; studies with random dot stimuli that minimize these confounds found no residual motion perception in 13 patients.176,177
Motion information might also guide eye movements. Recovery of optokinetic responses was reported in one patient with cortical blindness178 but not in two others168,179 nor in three hemianopic patients.174 Pursuit and saccadic responses to motion were not found in patients with medial occipital lesions sparing the lateral human motion area.137
An early report claimed that one patient could discriminate large X and O forms, perhaps through orientation perception.158,164 However, form discrimination was not found in seven other patients,155,163,180 nor orientation discrimination in two others.181,182 Despite this, three patients could arrange the orientation of their grasp correctly when they reached for objects in their blind field.159,181,183 This may be consistent with evidence for a dissociation between pathways for object recognition and action.184
Although early studies found no evidence of chromatic perception,155,164 later studies showed detection of colored targets185 and evidence of color-opponent interactions in the spectral sensitivity curves.186 One patient can perceive the motion of equiluminant colored spots187 and is aware of hue, though these responses seem to represent an average from the entire blind hemifield.188
Studies of temporal and spatial contrast sensitivity have yielded mixed results, even when done on the same patient GY.135,189,190
The standard pupillary light reflex that is clinically measured is a response to changes in luminance. However, there are also pupillary responses to gratings and isoluminant colors. Pupillary responses to such stimuli in the blind hemifield have been shown in one patient.191,192 In another study, color stimuli caused afterimages that evoked pupillary responses in normal subjects; in the blind hemifield of two patients, there were “after” pupillary responses without any conscious afterimage.193
A direct pathway between the pulvinar and the amygdala may mediate reactions to frightening stimuli.194 One patient could process fearful and angry faces in his blind hemifield195 and this may correlate with activity in his amygdala.196 Another study of a cortically blind patient showed that associating a visual stimulus with a painful shock caused the development of startle reflexes to that stimulus, despite the lack of conscious perception.197
INTERACTIONS BETWEEN BLIND AND NORMAL HEMIFIELDS.
Traditional blindsight methods are awkward in that they ask patients to respond to something they cannot see. However, a number of innovative studies have circumvented this by examining how responses to visible stimuli might be modified by stimuli in the blind field.
Spatial summation occurs when two simultaneous stimuli generate faster responses than a single stimulus; temporal summation is the decrease in reaction time when a stimulus is preceded by another that provides a temporal prompt. Evidence for summation between seeing and blind fields has been inconsistent and only found in a minority of subjects.167,198,199,200 The opposite, a distraction effect, in which targets in the blind field slow down response times to stimuli in the seeing field, has been shown for saccades but not manual reaction times.201 This finding could not be replicated in another study.202 Inhibition of return is a normal phenomenon in which a stimulus delays the detection of a target appearing in the same location a short time later. This phenomenon was generated in the blind hemifield of one patient.203
Word perception can be influenced in blindsight. The choice of meaning of an ambiguous word (i.e., LIGHT) in the seeing field can be influenced by a word in the blind field (i.e., DARK versus HEAVY).159 Completion effects have been reported for form perception. Two patients had completion effects for afterimages or with illusory contours such as the Kanisza triangle.159 The width of one patient’s grasp as he reached for an object varied with object size only when the left-most part of the object fell in his seeing hemifield.183 Another study used an interference task with a stimulus flanked by distractors either different or identical to the stimulus. Reaction times to seen letters and colors were prolonged by differing flankers placed in the blind field in a patient with an occipital lesion.204 Finally, one study tested whether perception of optic flow within the intact hemifield could be enhanced by optic flow in the blind hemifield; this could be shown in two patients with very limited damage to striate cortex199 (Fig. 15D).
BLINDSIGHT AND HEMIDECORTICATION.
Patients lacking a cerebral hemisphere are of interest in the debate over whether blindsight requires extrastriate cortex. Some have found that hemidecorticate infants will look toward their blind field when a target is presented there,205 and adults with such lesions have some residual manual localization of blind field targets.169,171 Also reported are motion and form perception169,171,206 and a spatial summation effect, in which patients responded faster to a seen flash when there was another simultaneous flash in the blind hemifield.207 However, the validity of these findings is in question. Some have failed to find blindsight outside of a narrow strip along the vertical meridian, which could be explained by receptive field overlap or light scatter.208,209 Other studies have also shown that the residual vision in hemispherectomized patients can be attributed to light scatter.210,211,212,213
ARTIFACT AND BLINDSIGHT.
Valid blindsight results require careful elimination of artifacts as alternative explanations of better than expected performance. Four issues need special consideration. First, poor fixation may allow the subject to place the target in the seeing hemifield. Eye position monitors can help, as long as both head and eye position are controlled or detected, since gaze direction is a function of both.214 Second, light scatter may diffuse from blind-field stimuli into the seeing field and mimic blindsight.137,210,211,212,215 Controls for scatter include testing the physiologic blind spot,158 or testing control patients with pregeniculate lesions 162,163,169,204 or hemispherectomies.174,212 Flooding of the seeing field with light has also been used to minimize scatter. Third, careful perimetry may reveal that the blind field has surviving islands of vision that account for the residual perception.216,217,218,219 Fourth, blindsight may simply represent a “criterion shift.” Subjects may reply more liberally when asked to choose between alternatives than when asked if they see something: this has been shown for normal subjects.215,220,221 Signal detection analysis has been used to answer this criticism,185,222,223 though it has also suggested that changes in criterion bias may indeed explain some blindsight motion perception results.224
EXPLANATIONS OF BLINDSIGHT.
Several different anatomic pathways are invoked to explain blindsight. Spatial localization may be accounted for purely by a subcortical pathway from the retina to superior colliculus.162,208 It is claimed that better blindsight in the temporal than nasal hemifield of patients is a signature of collicular mediation.201,225 Pattern or motion detection may require extrastriate cortex, through a relay involving the superior colliculus and pulvinar. Since the colliculus lacks color opponency, chromatic blindsight185,186,188,204,226,227 may indicate yet another relay, perhaps direct projections from surviving LGN neurons to extrastriate cortex.228
The monkey evidence most strongly supports the retino-tecto-pulvinar relay to extrastriate regions, particularly those involved in motion perception. Lesions of V1 do not abolish responses in V5229,230,231 or V3A232 unless accompanied by lesions of the superior colliculus.229,233 Stabilization of an early direct projection from LGN to V5, which normally regresses with development, may occur in infant cats with striate lesions,234 but evidence of this has not been found in infant monkeys.235
Physiologic techniques in humans have also provided some support. In normal subjects, evoked potentials236 and some transcranial magnetic stimulation studies237 but not others238 suggest that visual motion signals may arrive in V5 before and independent of V1. Positron emission tomography (PET) scans,154 magnetoencephalography,239 and evoked responses240 have shown residual activation of V5 by rapid but not slow moving stimuli in the blind hemifields of one patient. An intriguing functional MRI (fMRI) study of GY that varied stimuli to obtain responses with and without awareness showed that residual vision was associated with activation of extrastriate visual areas and dorsolateral prefrontal cortex, and blindsight with activation in superior colliculus and medial prefrontal cortex.160
Theories of blindsight must also account for its variability. Recent large series, representing 46 patients in total, suggest that blindsight is rare.176,217,218 One important variable may be the extent of additional damage to the optic radiations and extrastriate cortex. However, correlations between blindsight abilities and lesion anatomy have proven elusive.137,165,176,198,241 A requirement for very focal striate damage is also difficult to distinguish from a need for partial striate damage,199 pointing back to a potential artifactual explanation. Another important variable may be the timing of the lesion. Blindsight may require neural plasticity. If so, age at onset, time since lesion, and possibly training may be important.157 Infants or children may be more likely to develop blindsight or residual vision in both nonhuman primates and humans.155,169,234,242 Some reports imply that up to half of all children with cerebral blindness may have some residual vision for moving objects.243 Not all studies find that age matters, however.206 The efficacy of training in eliciting blindsight is controversial with both negative155,214 and positive170,241,244,245,246 opinions.
DISORDERS OF THE OCCIPITO-TEMPORAL PATHWAY
Disorders of Color Processing
Cerebral dyschromatopsia.
Cerebral (central) achromatopsia refers to complete loss of color perception, whereas cerebral dyschromatopsia indicates some residual color perception, which is the more common scenario. Both are rare. Hemiachromatopsia refers to color loss restricted to the contralateral hemifield247,248 and may be more common but under-recognized.
Achromatopsic patients are generally symptomatic, complaining that the world appears in shades of gray.249,250,251,252 Some also report that the world appears less bright253 or has a “dirty gray” tinge.254 Less frequently, patients report a tinge to the world, as if peering through a colored filter.255 Daily activities that use color discrimination are impaired, such as distinguishing coins, stamps, or traffic lights; a good account of the experience of an achromatopsic artist exists.256
Effects on color constancy are an important issue. The wavelengths reaching the eye from an object depends upon both its reflectant properties and the light illuminating the scene. Yet, the color of objects remains stable under a wide range of environmental and lighting conditions.257,258 For instance an apple continue to looks red in sunlight, incandescent light and fluorescent light, in an orchard or a grocery display. This ability to “discount the illuminant” depends on neural computations in retina and cortex.257 These computations likely average the spectral luminance over large regions of the surrounding background to deduce the nature of the illuminant, and this information is then taken into account to derive the true color of any object in the scene.257,259
A defect in color constancy should result in color percepts that vary with changes in illumination. Patients with achromatopsia have a more severe deficit, in that they lack any color perception at all. Testing such patients for constancy of something they do not perceive is paradoxical, but this can be done on dyschromatopsic patients who have some residual hue sensitivity. These studies have shown that color constancy is indeed impaired in these patients,260,261,262,263 all of whom had bilateral lingual and fusiform gyral lesions, except for one unilateral case.261
Not all color perception is lost in achromatopsia. Some color processing of the cones and retinal ganglion cells of the parvocellular pathway can still be discerned in testing. Both trichromacy and color opponency have been shown in photopic spectral sensitivity curves260,264,265 and evoked potential or psychophysical measures of chromatic contrast sensitivity.265,266 Likewise, performance with anomaloscope testing can resemble an anomalous trichromat rather than a monochromat, despite the experience of the world as monochromatic shades of gray.267 Achromatopsics can use color-opponent signals to detect a difference between colors and locate boundaries between differently colored regions of an image, even though they do not know what the colors are and cannot order them correctly by hue. This can help some patients detect the movement of chromatic stimuli,268,269 even performing at normal levels with suprathreshold chromatic contrast,270 indicating that wavelength variation is still perceived, even if color is not.
Achromatopsia is seldom an isolated finding. Most commonly it forms one component of a tetrad that includes superior quadrantanopia, prosopagnosia, and topographagnosia. Superior field loss is almost always present, as the ventral occipitotemporal lesion that causes achromatopsia frequently extends into the inferior calcarine cortex or optic radiation. Similarly, only a few cases without prosopagnosia have been reported.271 Experimental testing has revealed in some patients a problem with detecting stimuli with low salience,272 which has also been described in monkeys with V4 lesions, and thought to indicate inefficient attentional allocation in form processing. Other occasionally associated defects include visual object agnosia,264,273 alexia in achromatopsic patients with right hemianopia,251,271 and amnesia with additional ventral temporal lobe damage.251,273
Achromatopsia is caused by lesions of the lingual and fusiform gyri,258,274 as confirmed by modern imaging.253,254,264,267,271,275 Lesions of the middle third of the lingual gyrus or the white matter behind the posterior tip of the lateral ventricle may be critical,253,276 as suggested by a lesion meta-analysis.277 Bilateral lesions are necessary for complete achromatopsia.
In monkeys, color-selective responses are found in area V4.258 However, lesions of V4 do not impair hue perception significantly.278,279,280,281,282,283 Rather, defects in chromatic perception require extensive bilateral lesions, including areas TE and TEO.284,285 In humans, functional imaging reveals several areas involved in color processing, notably a V4 homologue and a second area named V4 alpha or V8 in the fusiform gyrus,286,287 as well as other more distant regions.288,289,290 Thus color processing involves a network of regions, and it is probable that a severe achromatopsic defect may require damage to or disconnection of several components of the network, rather than just a lesion of a single region like the human V4 homologue.281,291,292
Achromatopsia is most often due to strokes. Bilateral sequential or simultaneous infarctions in the territories of both posterior cerebral arteries can occur, or multiple infarcts may be due to a coagulopathy.293 Achromatopsia may be the first symptom of a stroke or the final outcome from an initial cortical blindness. Other bilateral lesions causing achromatopsia include herpes simplex encephalitis,264 cerebral metastases,271 repeated focal seizures,294 focal dementia,295 and even migraine aura, causing a transient achromatopsia.296 Temporo-occipital white matter damage has caused a reversible dyschromatopsia in one patient with carbon monoxide poisoning,297 a condition that usually causes an apperceptive agnosia with spared color perception.258
HEMIACHROMATOPSIA.
Achromatopsia in the contralateral hemifield alone can follow unilateral right or left occipital lesions (Fig. 15E). Patients are typically asymptomatic until the defect is demonstrated upon examination.247,248 Hemiachromatopsia is usually associated with a superior quadrantanopia,247,248,254 therefore, the color defect is only demonstrable in the remaining inferior quadrant. The preserved color vision in the ipsilateral hemifield allows normal or near-normal performance on centrally viewed tests of color vision such as pseudoisochromatic plates. The incidence of hemiachromatopsia is probably underestimated, given its asymptomatic nature and the failure of routine clinical color tests to detect its presence.
Two rare cases with quadrantic field defects for color have also been described.298 It is not clear if these were true chromatic defects or subtle relative scotomata, but the quadrantic representation of the human V4 area on functional imaging292 suggests that a very discrete lesion could theoretically create such a defect.
TESTING ACHROMATOPSIC PATIENTS.
Color naming is not an adequate test for cerebral dyschromatopsia, since residual color perception may allow an approximate categorization of colors. The deficits in color perception in the full syndrome of cerebral achromatopsia can be tested with the same standardized tests used to assess patients with retinal and optic nerve disorders.299,300,301 Pseudoisochromatic plates such as the American Optical Hardy-Rand Rittler302 and Standard Pseudoisochromatic Plates, Part 2303 are useful, even in patients with alexia or aphasia, who can do this test by tracing perceived patterns with their fingers. However, some patients with achromatopsia may still pass this test, particularly if the plates are presented so distant that the individual color dots cannot be resolved.251,264,275,303
Color arrangement tests consist of color chips mounted in caps that can be arranged in a unique sequence. They vary in the number of chips, the difficulty of the discriminations required, and the dimension of color space they probe, namely hue, saturation, or brightness. The Farnsworth-Munsell 100 Hue evaluates hue discrimination of tokens that do not vary in luminance. The D-15 test is a shorter version that screens for severe hue discrimination loss along protan, deutan, or tritan axes. The Lanthony New Color Test similarly tests hue discrimination, but at different saturation levels, and it also asks which colors are confused with grays. The Sahlgren Saturation Test304 evaluates saturation discrimination by testing the ability to separate five greenish-blue and five bluish-purple caps of varying saturation. The Lightness Discrimination Test consists of caps of different grays, to be ranked from dark to light.305,306 Achromatopsic patients often have abnormal discrimination of hues and saturation264,275,307 but normal perception of brightness.252,275,307
Anomaloscopic techniques have also been applied to study cerebral achromatopsia.253,267 With the Nagel anomaloscope, the observer tries to match a yellow monochromatic light in one test field with a mixture of yellow-green and yellow-red lights in another, by varying the proportion in the mix. Normal observers rapidly find the unique proportion needed, whereas individuals with congenital color defects or acquired cerebral achromatopsia lack a unique solution but have abnormal matches over a wide range.
The achromatopsic abnormality affects all hue perceptions diffusely, unlike that of congenital color blindness, although the magnitude of the defect along red-green and blue-yellow axes may vary relatively.253 The degree of achromatopsia can vary between patients, from complete to partial defects. Functional imaging studies suggest that this may depend upon the extent of damage to not just one but a number of color processing regions in occipitotemporal cortex.308
Hemiachromatopsia is more difficult to test. Hemifield color loss can be detected by having patients report on the appearance of large color tokens moved from the ipsilateral to the contralateral hemifield. For example, a red pen may appear to turn grayish to the patient as it is moved into the aberrant field. Yet it is difficult to quantify the color loss because the standard color tests above are designed for viewing within the central few degrees of vision where color vision is best. Hemiachromatopsic patients can achieve normal color scores on these tests because of spared color vision near fixation.253
Color anomia and agnosia.
Some patients cannot recognize or name colors, even though they can perceive them. Thus, while patients with achromatopsia cannot discriminate hue and saturation, though some can name some colors, those with color anomia and color agnosia can discriminate colors accurately but not name them. Such patients may not be aware of their deficits.
Color anomia may occur as part of a more general anomia in aphasic patients, or as an isolated entity. Several types of specific color anomia have been described, all with left occipital lesions and often an associated right homonymous hemianopia, rather than the upper quadrant defects of achromatopsia. When it accompanies pure alexia and right homonomous hemianopia, color anomia may be part of an interhemispheric visual-verbal disconnection.309,310,311,312 Disruption of callosal connections in the splenium between the intact right striate cortex and the left angular gyrus prevents accurately perceived colors in the remaining left hemifield from gaining access to language processors. Thus these subjects cannot read words or name colors that they see accurately in the left hemifield. On the other hand, some but not all of these subjects can name visual objects. Preservation of object naming has suggested that, unlike colors, objects can activate not only visual but also somesthetic representations of object shape, and that these representations could be transferred to the left hemisphere through more anterior corpus callosum.310 This remains to be proven.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


