Retinopathy of Prematurity
James D. Reynolds
RETINOPATHY OF PREMATURITY (ROP) is a complex disorder of the developing retinal vasculature in the immature retina of prematurely born infants. A relatively harmless and spontaneously resolving disease in the majority of affected infants, it unfortunately can threaten to cause blindness in a significant minority and even today can still result in blindness despite the best medical care.
ROP was first described in 1942 by Terry. His initial, brief report referred to this new disease as possibly a “fibroblastic overgrowth of the persistent tunica vasculosa lentis” (1). His second report was more extensive and involved several colleagues, one of whom, Messenger, formulated the descriptive term “retrolental fibroplasia” (2). Although not completely endorsed by Terry, the name stuck and the disease was known as retrolental fibroplasia (RLF) for the next 40 years. The second report, containing several pathologic specimens in the cicatricial end stages of the disease with multiple complications, clearly misinterpreted the involved pathophysiology. Terry did not recognize the retina as the source of the problem and still linked it to the rare congenital birth defect of persistence of the hyaloid artery and tunica vasculosa lentis. But he did recognize the salient epidemiology— that “some new factor had arisen” and the condition was occurring much “more frequently in infants born extremely prematurely.”
Terry concluded the initial case report with an entreaty to determine the “frequency, cause, and full nature” of the condition to discover “prophylactic treatment and effective therapy.” More than half a century of intense clinical and laboratory research efforts have made great strides in developing “effective therapies” for severe stages of ROP, and the understanding of the “nature” of this condition has improved tremendously. But little progress has been made in truly understanding the “cause” of this disease and even less in developing effective “prophylactic treatment.” The central mysteries of this disease have not been adequately unraveled to effectively eradicate unfavorable visual outcomes.
Individuals with ROP-induced blindness are far fewer than those afflicted with blindness from macular degeneration or diabetic retinopathy. Nonetheless this is a significant disease to the affected children, their parents, and to society in general. Steinkuller and coworkers (3) analyzed childhood blindness in the 10 years preceding their 1999 report. They found the three leading causes of pediatric blindness in the United States were cortical visual impairment, ROP, and optic nerve hypoplasia. Cortical visual impairment results from a variety of brain insults, often in utero, and usually is associated with other global sequelae. Optic nerve hypoplasia is a birth defect in fetal development without a known cause. Neither condition presents an opportunity for treatment. Thus, the leading cause of preventable blindness in children in the United States is ROP. This ongoing problem of ROP-induced infant blindness in the United States is confirmed by the Blind Babies Foundation (4).
Expanding the scope outside the United States finds a worldwide epidemic rivaling the U.S. experience in the 1940s and 1950s. Gilbert and coworkers (5) analyzed ROP throughout the world. They divided the world into three groupings of ROP epidemiology, which correlated highly with national wealth. In high-income countries like the United States, many extremely premature infants are saved, effective ROP treatments are universally applied, and ROP-induced blindness occurs but is limited. In low-income countries like most of those in Africa, extremely premature infants simply do not survive due to a lack of intensive care nursery technology. Hence, ROP blindness does not exist. However, in middle-income countries, such as in Latin America or Asia, intensive care is available, premature infants survive, but adequate means for screening and/or treatment is not available and ROP blindness is epidemic. The World Health Organization (WHO) and various partnering agencies launched the VISION 2020 program, which targeted childhood blindness and confirmed the need for ROP services, especially in middle-income countries (6).
One such middle-income country is Vietnam. A 1-year prospective series at a single maternity hospital in Ho Chi Minh City quantified the assessment of ROP risk (7). This study demonstrated an incidence of any ROP similar to that in the United States. But the incidence of severe ROP was considerably greater, and ROP was present in larger, older babies. Unfortunately, it also demonstrated a high rate of blindness resulting from less-than-ideal management.
In addition to the quantity of the problem, the socioeconomic costs relate to the quality of the problem. Apart from
the number of individuals affected, there is a qualitative difference between a lifetime of blindness and end-of-life blindness. Gilbert and Foster refer to this in terms of “blind years” and state that the worldwide number of blind years resulting from childhood blindness is similar to the quantity of blind years resulting from adult cataracts (8).
the number of individuals affected, there is a qualitative difference between a lifetime of blindness and end-of-life blindness. Gilbert and Foster refer to this in terms of “blind years” and state that the worldwide number of blind years resulting from childhood blindness is similar to the quantity of blind years resulting from adult cataracts (8).
Another major socioeconomic issue relates to the form of ROP management. Our current understanding of ROP has limited any effective prevention. Treatment regimens have been limited to high technology, high skill, and high-cost interventional therapy. This makes treatment expensive in high-income countries and unaffordable in middle-income countries. This has been changing with the use of intravitreal injections of bevacizumab.
HISTORY
The history of ROP in its early years as a public health epidemic and the scientific investigation involved in the search for answers is fascinating and holds lessons that are still relevant today. This disease seemed to come from nowhere when it burst on the nursery scene in the 1940s. As noted above, it was first described by Terry (1,2). Following this, RLF developed into a full-blown epidemic, blinding 10,000 babies between 1942 and 1954 (9). A frantic search for cause and effect and potential management was initiated. The only sure fact was that RLF occurred with an alarming frequency in premature infants whose lives were being saved for the first time by new approaches to caring for such infants in technologically advanced nurseries. But investigators were beginning to suspect supplemental oxygen administration (10,11).
Jacobson and Feinstein performed a postmortem on the clinical epidemiologic research attempt to solve the RLF puzzle (9). The authors painstakingly described and analyzed a “decade of errors” in this research. Poor methodology, misleading assumptions, reliance on small sample size, empirical methods, investigator bias, lack of controls, and lack of randomization all contributed to dead ends. The story culminates in the success of the multicenter, randomized clinical trial, the National Cooperative Study, in definitively uncovering the correlation between increased exposure to supplemental oxygen and RLF (12,13). These supplementary oxygen revelations resulted in a curtailing of limitless oxygen administration in the nursery. An oxygen policy based on the least amount of inspired oxygen required for survival dramatically lowered the incidence of blind babies. But it did not eliminate them. And unfortunately, it did not minimize mortality and morbidity (14). The oxygen controversy, thought to be put to rest in 1955, still rages today. Silverman states in an editorial that “there has never been a shred of convincing evidence to guide limits for the rational use of supplemental oxygen in the care of extremely premature infants” (15).
In the last half century, in the continuing quest to answer the questions posed by Terry (1), researchers did not always learn from their mistakes. False passages illuminated by poor methodology continued to plague ROP research. Resurrection of perhaps prematurely discarded ideas came into vogue. It required the age of the multicenter randomized trial to arrive in conjunction with basic laboratory investigation to begin to characterize ROP. The rest of this chapter will concentrate on the last 25 years of ROP research, defining what is now known about Terry’s “nature, cause, prophylaxis (sic), and therapy.”
CLASSIFICATION
An early difficulty in epidemiologic research was the lack of a universal classification of ROP. Investigators were not always speaking the same language, meaning that important elements of the disease could go unrecognized or unappreciated. This serious impediment was removed in 1984 with the publication of the International Classification of Retinopathy of Prematurity (ICROP) (16). This was the key to opening the door to rigorous trials. Its relevance was immediately recognized, and clinicians and researchers alike embraced the classification. Its significance cannot be overstated. Overnight, the epidemiology landscape changed dramatically.
Besides providing this crucial basis for further research, the classification had another, more subtle, much less recognized but still very important benefit. The process of its development and adoption brought many of the central investigators together. It served as a model of cooperation and collaboration and paved the way for the future multicenter trials that would so define this disease. So the classification provided both the scientific foundation and philosophic approach for subsequent investigations.
The core of the classification was defining the stages of the acute disease and presenting a topographic map upon which to locate the disease in the retina. Both have major clinical significance. Staging of ROP relied upon clearly definable and observable structural changes in the retina. The stages proceeded from mildest to most severe disease and were classified as stages 1 through 4. These stages were usually easily recognized by experienced examiners utilizing the indirect ophthalmoscope, which provides the important three-dimensional picture of this disease. The stages of ROP were agreed to be the following:
Stage 1: line of demarcation
Stage 2: ridge of elevated tissue
Stage 3: neovascularization with extraretinal fibrovascular proliferation
Stage 4: retinal detachment
4a: macula not involved
4b: macula detached
Stage 5: total retinal detachment
More descriptively, stage 1 is a circumlinear, whitish, thin, flat line distinctly separating normally vascularized
retina from as yet unvascularized retina. Stage 2 is present when this circumlinear line becomes thicker and more elevated and forms a true ridge extending out of the plane of the retina. The three-dimensional view of the indirect ophthalmoscope is crucial here. Stage 3 is extraretinal fibrovascular neovascularization. The fibrous component has a very different appearance than the neovascularization in diabetes or sickle cell disease. This neovascularization is more of a continuous sheet of solid pink tissue (Fig. 3.1). Fronds of individual vessels typical of other diseases are not seen in ROP. Stage 4 represents retinal detachment, small or large, shallow or high. The retinal detachment may be exudative, tractional, or both, but is not rhegmatogenous. Stage 5 is an addition to the International Classification used to denote a total retinal detachment, either open funnel or closed (17) (Fig. 3.2).
retina from as yet unvascularized retina. Stage 2 is present when this circumlinear line becomes thicker and more elevated and forms a true ridge extending out of the plane of the retina. The three-dimensional view of the indirect ophthalmoscope is crucial here. Stage 3 is extraretinal fibrovascular neovascularization. The fibrous component has a very different appearance than the neovascularization in diabetes or sickle cell disease. This neovascularization is more of a continuous sheet of solid pink tissue (Fig. 3.1). Fronds of individual vessels typical of other diseases are not seen in ROP. Stage 4 represents retinal detachment, small or large, shallow or high. The retinal detachment may be exudative, tractional, or both, but is not rhegmatogenous. Stage 5 is an addition to the International Classification used to denote a total retinal detachment, either open funnel or closed (17) (Fig. 3.2).
The topographic map of the retina devised by the International Classification is shown in Figure 3.3. It divides each retina into three zones. The goal of these divisions was to be clinically relevant, yet easily recognized. These zones are defined as follows:
Zone I: a concentric circle, centered on the optic nerve, with a radius of two times the distance from the center of the nerve to the center of the fovea
Zone II: diagrammatically a concentric annular area arising from the outer border of zone I and ending at the ora nasally and just beyond the equator temporally
Zone III: a large temporal crescent arising from the outer border of zone II and terminating at the temporal ora serrata
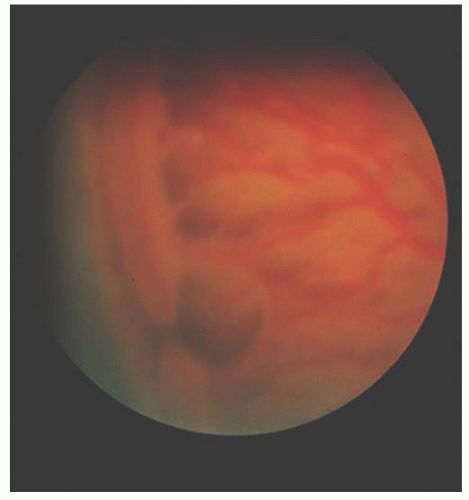 FIGURE 3.1. Typical, moderately severe stage 3 ROP. Note the continuous sheet of extraretinal, fibrovascular tissue. Even in this two-dimensional photo, the elevation is apparent. ROP, retinopathy of prematurity. |
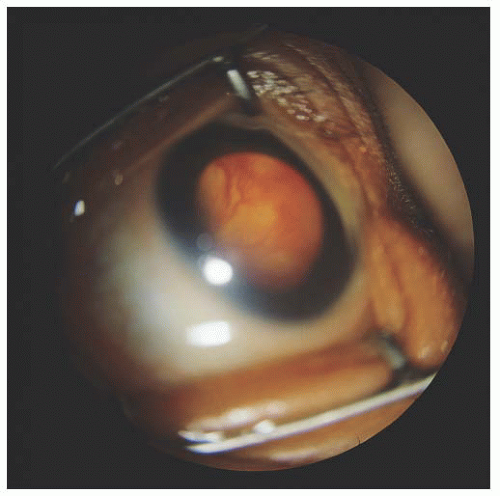 FIGURE 3.2. Stage 5 total retinal detachment with open funnel. |
There are several very important points to understand about the location classification that have bearing on the way the natural history of this disease plays out on this topography. First, it is an arbitrary classification. The zones were picked to enhance ease of recognition. The optic nerve, macula, and nasal and temporal ora are all distinct and identifiable by indirect ophthalmoscopy. Recognition of the equator, which can be difficult, is irrelevant to the classification. Secondly, the macula is the true anatomic center of the eye, not the optic disc. But vascularization of the retina proceeds centrifugally from the optic disc to the ora. Since the optic disc is in the nasal retina, normal vessel development reaches the nasal ora first, leaving the as yet unvascularized temporal crescent. If observed at just the right moment in time, this temporal crescent can actually extend to well over 300 degrees of the peripheral retina. Thirdly, there is no defined border between zones II and III on the temporal side. Because there is no easily identifiable midperipheral landmark to use as a reference, the temporal location is defined exclusively by the relationship of the normal vessel growth or ROP pathology to the nasal ora. No matter where the real location is temporally, if the vessels have not reached the nasal ora, the location is zone I or II. If the vessels have reached the nasal ora, then the location is defined as zone III. With normal, uninterrupted vascularization, the diagrammatic representation of a concentric zone II reflects reality. But when pathology supervenes, it is possible to have temporal disease in zone II that does not physically move but becomes arbitrarily classified as zone III when nasal vascularization is less impeded by pathology. In other words, nonconcentric ROP can exist in the presence of asymmetric location of disease onset, progression, or involution.
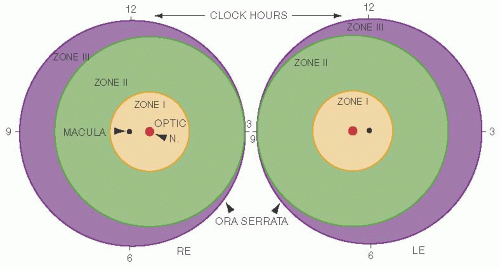 FIGURE 3.3. Schematic representation of the retinas divided into three zones, with the relevant anatomic landmarks. (Reprinted from the Committee for the Classification of Retinopathy of Prematurity: an international classification of retinopathy of prematurity. Arch Ophthalmol 1984;102:1130-1134. Copyright American Medical Association, 1984. All rights reserved.) |
Although minor, it is a flaw in the classification that the three zones reflect the natural progression of normal vascularization but do not always accurately reflect the true location of temporal disease. A true representation would relate temporal disease to its anatomic proximity to the macula in zone II as it does in zone I. This would realistically predict its ability to impact the macula and central vision. But since temporal disease location is defined by nasal pathophysiology, temporal disease is always, at best, an estimated location. As will be shown in subsequent sections, this has an impact on natural history and screening guidelines. In summary, the location classification is grounded in normal physiology of vascularization, is easily identifiable, and is highly clinically relevant.
By convention, ROP is clinically classified by the highest stage and lowest zone. The retina is imaginarily divided into twelve equal radial segments, commonly referred to as clock hours, and this represents the extent of the disease. Disease can be present for as little as 1 clock hour or as many as 12. But the highest stage and lowest zone of just 1 clock hour determines the disease classification at that time. For example, 1 hour of stage 3, zone I and 11 hours of stage 2, zone II is diagnosed as stage 3, zone I disease.
Finally, the concept of plus disease was introduced (16). Plus disease refers to posterior pole large vessel engorgement and tortuosity. Plus disease occurs in response to a stimulation for increased blood flow. There is usually a neovascular shunt present, which itself has arisen due to ischemia signals within the retina. And plus is more likely to occur when the ROP is more posterior, possibly because the large vessels respond more to an anatomically nearer ischemic microenvironment. Along with flagrant plus disease comes vitreous haze, iris vessel engorgement, and iris resistance to mydriatics. This hemodynamic change is a threshold concept that requires a minimum degree of change before it can be termed plus. Its specific definition is:
Plus disease: posterior venous dilation and arteriolar tortuosity of at least a photographically defined minimum
But the process of developing plus disease is not allor-none. Posterior pole vessel dilatation and tortuosity can be of gradual onset or to a level not recognized as full plus. Thus, plus disease requires the observer to make a quantifiable judgment of an inherently qualitative clinical sign. This is far different from the other portions of the classification. Stage and location are not a question of degree. Often this judgment is easy with flagrant plus disease being obvious. But borderline plus disease can be difficult. The very word borderline implies the difficulty involved. The minimum level of plus disease required has been traditionally taught to ROP examiners by way of a single standard photograph (16). It has never been put into a prose definition and never been objectively quantified. Various investigators have subdivided pre-plus changes, but they have not been widely accepted (18,19,20). Additional work has highlighted interobserver disagreement, variability in the nature of dilation versus tortuosity, and the potential of computer-assisted photographic modeling systems (21,22,23,24).
Another difficulty in judging plus disease is the impact that the examination has on the clinical picture. Scleral depression can impede blood flow by temporarily raising the intraocular pressure. Reduced blood flow results in reduced dilation. Prolonged examination, perhaps by multiple, sequential observers, can increase blood flow, a kind of rebound effect. Again, a crucial judgment is made more difficult and the observer must be cautious.
The ICROP II described and classified retinal detachments, as well as regression patterns of ROP. Special attention was given to peripheral versus central changes. Regression of acute ROP is synonymous with the onset of cicatricial disease development (17).
The International Classification, parts I and II, was an enormous step forward. It has an ease of use that allows reliable interobserver assignment of disease stage, location, extent, and presence or absence of plus disease. Equally important is its strong clinical relevance. The observer needs only some experience and an awareness of the above caveats.
ICROP was revisited in 2005 (25). This resulted in the addition of “pre-plus” and “AP-ROP.” Pre-plus indicates a degree of vascular dilation and/or tortuosity below the minimum needed for plus disease designation. This is an important prognostic sign. AP-ROP is acute posterior ROP, and it describes an aggressive form of ROP that is rapidly progressive and is located in zone I or posterior zone II. It tends to have an earlier onset and can advance to treatable disease surprisingly quickly. This replaces the old term of “rush disease.”
Subsequent subsidiary classifications have become clinically universal. These do not alter or depart from the International Classification but have added clinically relevant subclassifications. The Multicenter Trial of Cryotherapy for Retinopathy of Prematurity (CRYO-ROP) developed the concept of assigning clinical significance to a certain level of disease (26). Although arbitrary and developed as a definition to determine study intervention, the concepts proved so clinically useful that they were adopted to define the standard of care. These two ubiquitous definitions are prethreshold ROP and threshold ROP. Prethreshold was defined as a near intervention level of disease severity, and threshold, as the name implies, was a level of disease severity which triggered study intervention. These definitions are:
Prethreshold ROP: any stage ROP in zone I, stage 2 zone II plus, or any stage 3
Threshold ROP: at least 5 contiguous or 8 cumulative clock hours of stage 3 zone I or II with plus
These two concepts are ingrained in clinical parlance, and the terms are always used to mean the exact definitions above. They no longer define the standard of care for disease intervention, but they did so for 15 years.
The Early Treatment of ROP (ET-ROP) trial revisited the concept of threshold ROP and redefined a new treatment standard, replacing threshold (27). Type I ROP became the new intervention point and type 2 ROP designated pre-intervention disease. These are specifically defined as:
Type I ROP: Zone I, any stage ROP, with plus disease, Zone I, stage 3 ROP, without plus disease, Zone II, stage 2 or 3 ROP, with plus disease.
Type 2 ROP: Zone I, stage 1 or 2 ROP, without plus disease. Zone II, stage 3 ROP, without plus disease.
Thus, the current clinical classification system utilizes the formally agreed upon International Classification and the clinically useful modifiers over the years and includes:
Stage
Location
Extent
Pre-plus
Plus disease
AP-ROP
Regression of disease (cicatricial pattern)
Prethreshold ROP
Threshold ROP
Type I ROP
Type II ROP
All examiners should be familiar and facile with these terms, their definitions, and most important, their clinical appearance and relevance.
SCREENING AND EXAMINATION
Screening for ROP is an essential element in the management of ROP. The cardinal rule is that the patient must be examined before the patient can be treated. The cardinal sin is failing to examine the patient within the window of treatment opportunity. Appropriate screening requires not only scientific evidence to outline the screening parameters but also appropriate administrative supervision in applying those screening parameters to each at-risk infant.
An ideal evidence-based screening protocol should detect serious disease in a timely fashion, consistently, reliably, and cost effectively. It should minimize the number of examinations required and maximize the opportunity for intervention. It does not have to account for every conceivable exception. In fact, screening programs by nature must have parameters based in population statistics, not in exceptional circumstance. Risk must be reasonably defined.
Examination of the entire premature infant retina requires a well-dilated pupil and indirect ophthalmoscopy with lid speculum and scleral depression. Minimizing examinations is especially important in premature infants under-going the stress of pharmacologic pupillary dilation and scleral depression. These tiny infants are especially medically unstable in the early weeks of ROP screening examinations. Reported complications include cardiopulmonary arrest, apnea, bradycardia, tachycardia, alterations in blood pressure, decreased oxygen saturation, inadvertent extubation, gastric reflux, and infection (28,29,30,31,32,33). In addition, unnecessary examinations add to the expense of care and may inconvenience families and expose infants to infection risk when forced to attend unnecessary outpatient examinations. Thus, ROP screening guidelines should provide for when to appropriately begin examinations, how often to examine, and when to conclude examinations. In addition, the at-risk population needs to be reasonably defined.
Screening guidelines have been developed and updated many times from many sources. These sources include single-center experience, as well as political consensus documents. Utilizing these recommendations, a reasonable conclusion as to the at-risk population would be all premature infants with a birth weight of less than or equal to 1,500 g or with gestational ages of 31 weeks or less. Keep in mind that this represents the at-risk population in high-income countries. The Vietnam series would suggest that larger, older babies are at risk in middle-income countries (7). This is probably due to differences in the standard of care available in the neonatal intensive care nurseries.
The timing of ROP screening no longer needs to rely on single-center data or consensus policy statements. Reynolds and coworkers utilized the databases from the CRYO-ROP study (34) and the Light Reduction in Retinopathy of Prematurity (LIGHT-ROP) study (35) “to define appropriate ages and retinal ophthalmoscopic signs for the initiation and conclusion of acute-phase ROP screening” (36). The goal was to examine the infant within the window of opportunity for ideal treatment intervention while minimizing exams. An additional goal was to provide for an evolving definition of the ideal point of intervention in the disease spectrum without requiring alteration of the guidelines. Figure 3.3 shows the basic data used by the authors, and Figures 3.4 and 3.5 demonstrate the near identity of CRYO-ROP and LIGHT-ROP despite a decade of separation. This identity of disease
onset validates the applicability of CRYO-ROP data to today’s situation.
onset validates the applicability of CRYO-ROP data to today’s situation.
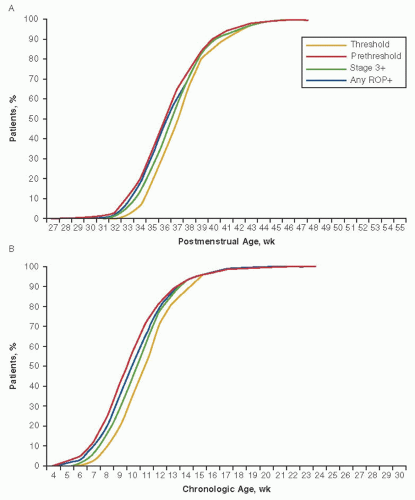 FIGURE 3.4. Timing of the onset of threshold, prethreshold, stage 3 plus, and any ROP with plus disease by postmenstrual age (A) and chronologic age (B). ROP, retinopathy of prematurity. (Reprinted from Reynolds JD, Dobson V, Quinn GE, et al. for the CRYO-ROP and LIGHT-ROP Cooperative Groups. Evidence based screening criteria for retinopathy of prematurity. Arch Ophthalmol 2002;120:1470-1476. Copyright American Medical Association, 2002. All rights reserved.) |
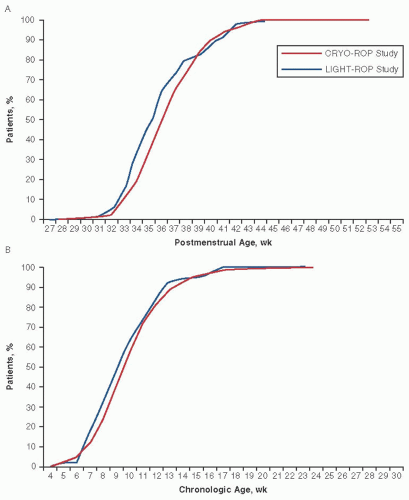 FIGURE 3.5. Timing of the onset of prethreshold ROP for CRYO-ROP and LIGHT-ROP patients less than 31 weeks’ gestational age at birth by postmenstrual age (A) and chronologic age (B). Note the nearly identical curves. CRYO-ROP, cryotherapy for retinopathy of prematurity; LIGHT-ROP, light reduction in retinopathy of prematurity; ROP, retinopathy of prematurity; (Reprinted from Reynolds JD, Dobson V, Quinn GE, et al. for the CRYO-ROP and LIGHT-ROP Cooperative Groups. Evidence based screening criteria for retinopathy of prematurity. Arch Ophthalmol 2002;120:1470-1476. Copyright, American Medical Association, 2002. All rights reserved.) |
Analyzing this data and more, the authors recommended that the timing of the initial exam follow the guidelines in Table 3.1. Based on accurate assessment of gestational age at birth, any infant can be plugged into the table, and the week of the initial exam can be definitively set in advance. Similar timing data, with the addition of prognostic retinal signs along with ROP involution data (37), can be used to determine a safe and appropriate time for the conclusion of screening. This work formed the foundation for the U.S. consensus paper published in 2006 and the most recent U.S. guidelines published in 2013 (38, 39, 40).
In summary, the following guidelines should determine screening for acute-phase ROP:
1. Subjects: premature infants less than or equal to 1,500 g birth weight or less than 31 weeks’ gestational age
2. Screening initiation: Table 3.1
3. Screening conclusion
a. Zone III retinal vascularization attained without previous zone I or II ROP, assuming no examiner error. If there is doubt about the zone or if the
postmenstrual age (PMA) is unexpectedly young, confirmatory examinations may be warranted.
postmenstrual age (PMA) is unexpectedly young, confirmatory examinations may be warranted.
b. Full retinal vascularization
c. PMA of 45 weeks and no prethreshold ROP or worse present
d. Definite disease regression signs in compatibly aged infants
Table 3.1 TIMING OF INITIAL EYE EXAM | ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||
In the section on ROP classification, a minor flaw in the system was noted that allowed temporal zone II ROP to be reclassified as zone III ROP without changing its true anatomic location. This is why the above screening conclusion guideline on zone III is qualified by excluding eyes with previous zone II ROP. These latter eyes probably represent most of the uncommon, unfavorable outcomes that occur with zone III ROP. In other words, the poor outcomes that rarely occur in zone III ROP probably had asymmetric temporal vs. nasal disease, and the nasal retina went on to vascularize while the temporal disease progressed without changing its real anatomic location. This is an important caveat for which the examiner must account.
Finally, although the initiation and conclusion of ROP screening exams in an appropriately at-risk population is now known, what about exam frequency and time to treatment? These latter two parameters are still consensus values arising from the conduct of multicenter trials. Exams are thought to be indicated every 2 weeks in infants with retinas showing stage 2 or less disease but should probably be weekly for plus or nearly plus disease, stage 3 disease, zone I disease, rapidly progressive disease, or disease occurring in an atypically young infant. Time from observation of treatable disease to application of treatment should be within 3 days.
This covers the scientific and disease-specific screening guidelines, but what of the administrative component? This is equal in importance. While the goal of evidence-based screening guidelines is to define the 99th percentile parameters, which appropriately exclude consideration of extraordinary events, the goal of the administrative component is the inclusion of 100% of the infants defined as at risk by the 99th percentile parameters. In other words, good science followed by good care. Such inclusive goals do not come easily or casually. All the interested parties must be involved in the attempt to set up a foolproof system of inclusion. Realistically no human activity is foolproof or perfect. But the goals should be set and clearly understood by all. System analysis is the operant process. It does not fall upon any nurse or any physician or any hospital employee to assure success. It is the system that is put in place, conscientiously adhered to by all, modified by the recognition and attempted correction of unforeseen events that will move toward perfection.
The involved parties include neonatologists, primary care pediatricians or family practitioners, nurses, social workers, discharge planners, ophthalmologists, quality assurance officers, clerical workers, and parents. Residents and fellows may also play a role but are appropriately peripheral in responsibility. No single party should accept full responsibility for ensuring inclusion. That is a recipe for disaster without checks and balances. Work as a team, institute the above scientifically based screening protocol, identify responsibilities and responsible parties, monitor progress, utilize system analysis, document the progress, communicate, and finally imbue all concerned with a sense of dedication and determination. The cost of failure is high, in more ways than one (40).
A useful mnemonic summarizes the above:
Delineate responsibility
Develop a process
Discuss frequently with all
Document the entire process
Do it
INCIDENCE
The prevalence of blindness from ROP in the United States appears to be growing despite effective treatments for severe ROP (3,4). The incidence of worldwide ROP blindness is unquestionably on the rise (5). But this could be related to a rise in the number of surviving premature infants and not to a change in the actual incidence or severity of ROP.
Several advances for the care of prematurely born infants have been instituted in neonatal intensive care units in high-income countries. These include surfactant administration, antenatal steroid use, pulse oximetry monitoring, improved nutrition, and others. Have these improvements in systemic care or other, as yet unknown, factors contributed to a change in incidence and severity of ROP as opposed to the prevalence of ROP blindness?
Some studies have suggested at least some type of decrease in ROP. However, these reports have serious flaws. The Vanderbilt experience compared 1995-1996 numbers with the years 1986-1987, when they were part of CRYO-ROP (42). It can be seen that the baseline years of 1986-1987 in Vanderbilt had threshold rates of 19% as compared with overall CRYO-ROP rates of 6%. The 1995-1996 numbers are much closer to both CRYO-ROP and LIGHT-ROP. Although the incidence of threshold ROP went down dramatically in Tennessee, the decrease was from an isolated exorbitant rate to a typical national rate. This study does not prove a decrease in incidence. Rather it proves the aberrations to which single centers with small numbers of babies are prone (43).
A study from an Australian center comparing data between 1988-1991 and 1991-1994 is subject to the same flaws (44). The threshold numbers were 25.0% versus 9.7%, respectively. The first number is extreme, and the second is still higher than the multicenter experience in the United States. This is also probably single-center aberration, but perhaps this is reflective of Australian nursery practices approaching U.S. standards. Conversely, a Danish study found no difference in ROP in high-risk infants (45). Termote and coworkers (46) reported a decrease in ROP in all patients, but an increase in those smaller than 1,000 g at birth.
No independent body of literature has addressed the effects of the previously noted medical advances on ROP incidence except for surfactant use. The prophylactic and therapeutic administration of exogenous surfactant to newborn premature infants has had a dramatic impact on lung disease and survival. Surfactant administration theoretically could increase severe ROP by increasing survival of low-birth-weight infants, decrease ROP by improving the general health of these infants, or impact ROP in other as yet unknown ways. Early studies found conflicting results.
The evidence now seems clear that surfactant administration does not reduce either the incidence or severity of ROP. In a study by Repka and coworkers (47), infants from two CRYO-ROP centers were evaluated in a randomized trial with prospective, serial eye exams. No difference in ROP outcome was noted. With this evidence, the authors postulated that the absolute number of babies with threshold ROP will raise as survival improves and birth-weight-specific incidence of ROP remains the same. Holmes and coworkers (48) also failed to find a statistically significant difference in their randomized trial.
The best way to assess the incidence and severity of ROP over time is to compare multicenter randomized trials. Despite technologic and medical advances in the intervening years, it appears that the rates of ROP are very similar. The CRYO-ROP study enrolled and followed 4,099 patients. Of those, 2,699 (65.8%) developed at least some ROP, 731 (17.8%) developed at least prethreshold ROP, and 245 (6.0%) developed threshold ROP (49). The LIGHT-ROP study enrolled and followed 361 patients. Of those, 251 (69.5%) developed at least some ROP (although only 202 developed the study’s minimum definition of at least 3 clock hours of confirmed ROP), 52 (14.5%) developed at least prethreshold ROP, and 18 (4.9%) developed threshold ROP (35). These two studies were conducted in 1986 through 1988 and 1995 through 1997. Table 3.2 shows that even though the enrollment periods were separated by almost a decade, the incidence and severity of ROP is very similar.
What of the more recent Early Treatment for Retinopathy of Prematurity (ET-ROP) randomized trial? Since this was a treatment trial that impacted the incidence of threshold, threshold ROP cannot be used for comparison. But the rate of prethreshold ROP could be used for comparison purposes (27). Utilizing these comparisons, the incidence and severity of ROP were very similar in all three multicenter trials. The one area of sizable difference is in the incidence of zone I disease. ET-ROP had a much higher incidence of zone I disease. While that may represent a true epidemiologic shift, it could also be at least partly explained by the differences in methodology as well as observer bias (50).
It seems safe to conclude that ROP remains a major health issue. The knowledge that ROP rates remain stable despite improvements in neonatal care has a major impact




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
