Retinoblastoma
Carol L. Shields
Jerry A. Shields
Retinoblastoma is the most common intraocular cancer of childhood.1,2 It represents approximately 4% of all pediatric malignancies. It is estimated that 250 to 300 new cases of retinoblastoma are diagnosed in the United States each year and 5,000 cases worldwide. Large countries such as India and China individually estimate approximately 1,000 new cases of retinoblastoma per year. Over 95% of children with retinoblastoma in the United States and other medically developed nations survive their malignancy, whereas about 50% survive worldwide. The reason for the poor survival in undeveloped nations relates to late detection of advanced retinoblastoma, often presenting with orbital invasion or metastatic disease. In Brazil, the mean age at presentation for retinoblastoma is approximately 25 months, compared with 18 months in the United States.3 Speculation is that Brazilian families delay seeking medical care for a mean of 6 months, and the delay is longer if the only symptom was strabismus of an eye with retinoblastoma (lagtime is 9 months) more so than symptoms of leukocoria (lagtime is 6 months) or tumor mass (lagtime is 2 months).3
Genetics: Basic Facts
Retinoblastoma affects approximately one infant in 15,000 to 20,000 live births in the United States each year.1,2,4 Most studies indicate that the incidence of retinoblastoma among the various geographic populations is relatively constant. The role of environmental influences in the development of this malignant intraocular tumor remains unclear. Before the 1860s and the role of enucleation in the management of retinoblastoma was known, most cases of retinoblastoma proved fatal. At that time, little was suspected about the inheritance patterns of this tumor because few patients survived to reproductive age. Later, as more patients survived and had children of their own, more evidence arose suggesting the hereditary nature of retinoblastoma.5,6 It is now known that retinoblastoma can be inherited as a familial tumor in which the affected child has a positive family history of retinoblastoma or as a nonfamilial (sporadic) tumor in which the family history is negative for retinoblastoma. Approximately 6% of newly diagnosed retinoblastoma cases are familial and 94% are sporadic. All patients with familial retinoblastoma are at risk to pass the predisposition for the development of the tumor to their offspring, but the manifestations are only 80% penetrant.
Retinoblastoma is generally classified in three different ways: (a) familial or sporadic, (b) bilateral or unilateral, and (c) heritable or nonheritable. From a clinical standpoint, the first two classification schemes are most often used.7 Thus, a case is classified as unilateral sporadic, bilateral sporadic, unilateral familial, or bilateral familial. About two-thirds of all cases are unilateral and one-third are bilateral. From a genetic standpoint, it is simpler to discuss retinoblastoma with the latter classification of heritable or nonheritable. The three classification schemes, however, are interrelated. It is recognized that bilateral and familial retinoblastoma are caused by a germline mutation and, thus, are a heritable tumor. Unilateral sporadic retinoblastoma is usually not heritable; however, it is estimated that approximately 10% to 15% of children with unilateral sporadic retinoblastoma have a germline mutation (Table 3-35.1). Genetic testing using DNA analysis of the patient’s tumor and peripheral blood can help identify those patients with germline mutation.8,9
Table 3-35.1. Retinoblastoma type and laterality | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Before the retinoblastoma gene was identified, linked markers such as esterase D were used to identify individuals who had the heritable form of retinoblastoma. Esterase D, an enzyme expressed in all cells, is coded on chromosome 13. If both of its alleles are fully active, then the enzyme is measured as 100% activity. If one allele is missing, then the activity drops to 50%. Esterase D has been found to be closely linked to the retinoblastoma gene on the 13th chromosome. In fact, it has been found to lie in the proximal portion of the 13q14 locus.10 Because of this tight association with retinoblastoma, previous screening for the retinoblastoma gene was done by testing for esterase D levels, but is rarely used today. If esterase D is 50% or less than its normal activity, then it suggests that a chromosome 13 deletion and possible retinoblastoma gene deletion is present.
The first clue to the location of the retinoblastoma gene was recognized by Stallard in 1962 when he reported the case of an infant with retinoblastoma who had a deletion in one of the D-group chromosomes.11 The D-group chromosomes include numbers 13, 14, and 15. It was difficult to distinguish these three chromosomes without the cytogenetic techniques that evolved in the 1960s. Additional cases of D-group deletion retinoblastoma were recognized and retinoblastoma became known as a part of the D-deletion syndrome. In the 1970s, high-resolution chromosomal banding showed that the affected chromosome in patients with retinoblastoma was chromosome 13.12 Because of this, the syndrome was renamed “the 13-deletion syndrome.” In 1978, the locus of the deletion was found to be the q14 band, which is the 14 band on the long arm (q) of the 13th chromosome.
The retinoblastoma gene is located on the long arm of chromosome 13 (13q14). It is a large 4.73 kilobase (kb) message. An intact gene protects against expression of retinoblastoma. It is believed that the gene is a recessive suppressor gene and may play a role in cell growth and development. For retinoblastoma to develop, both copies of the gene at the 13q14 locus must be lost, deleted, mutated, or inactivated. If either the maternal or paternal copy of the gene that is inherited by an individual is defective, then that individual is heterozygous for the mutant allele. Tumor formation requires both alleles of the gene to be mutant or inactive. These two mutations correlate to the two “hits” theorized by Knudson et al.,13 who in 1971, proposed the “two hit” hypothesis to explain the events that are necessary for both heritable and nonheritable retinoblastoma. This theory was based on a comparative analysis of unilateral and bilateral retinoblastoma cases and the proposal was that the development of any retinoblastoma was caused by two complementary chromosomal mutations. Each of these genetic events could occur randomly with a frequency of 2 × 10-7 per year. In the case of familial retinoblastoma, the initial event or “hit” was a germinal mutation that was inherited and found in all cells of the offspring. The second “hit” occurred sometime during development and, if it occurred in a somatic cell, such as a retinal cell, then retinoblastoma would develop. Therefore, in familial cases of retinoblastoma, all cells in the body are predisposed to possible tumor development because germline mutation (first hit) has been inherited in all cells of the body, including the ovaries and testes. This may help to explain the high incidence of second nonocular tumors, such as osteosarcoma, soft tissue sarcoma, and cutaneous melanoma, seen in patients with familial retinoblastoma or bilateral sporadic retinoblastoma. The offspring in cases of familial retinoblastoma are likewise predisposed because their germinal mutation will be passed on. By contrast, in most cases of unilateral sporadic retinoblastoma, the two hits occur during development of the retina and both hits are somatic mutations. The rest of the body theoretically carries no higher risk to develop other tumors because these patients presumably have normal chromosomal structure elsewhere in the body.
Genetics: 13q Deletion Syndrome
The retinoblastoma gene is located on the long arm of chromosome 13 (13q).14,15 The 13q deletion syndrome can manifest by several phenotypic abnormalities.8,9 Many patients have minimal or no visible abnormality.13 The characteristic findings include some degree of the following dysmorphic features: microcephaly, broad prominent nasal bridge, hypertelorism, micro-ophthalmos, epicanthus, ptosis, protruding upper incisors, micrognathia, short neck with lateral folds, large prominent low-set ears, facial asymmetry, imperforate anus, genital malformations, perineal fistula, hypoplastic or absent thumbs, toe abnormalities, and psychomotor and mental retardation.16,17 The midface of patients with a 13q deletion are notable for prominent eyebrows, broad nasal bridge, bulbous tipped nose, large mouth, and thin upper lip (Fig. 3-35.1). We reported a case of severe midline facial and central nervous system abnormalities in a child with 13q abnormalities that manifested retinoblastoma and holoprosencephaly.18
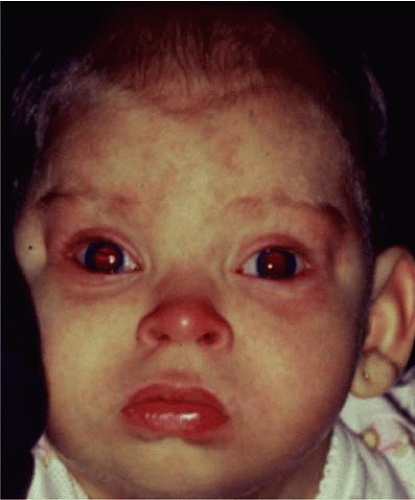 Figure 3-35.1. External appearance of a 12-month-old girl with 13 q syndrome. Note low set ears, bulbous tip of nose, and facial dysmorphism. She had a small paramacular retinoblastoma of the left eye. |
Karyotype analysis of children with these or other dysmorphic features may allow earlier detection of retinoblastoma. We have seen several cases of retinoblastoma that were initially suspected based on the recognition of the above dysmorphic features that prompted a karyotype analysis revealing a deletion in chromosome 13. This finding subsequently prompted a retinal examination which revealed unilateral multifocal tumors in both cases.17
Genetic Counseling Regarding Future Offspring
One of the more important steps in the management of the case of a patient with retinoblastoma is genetic counseling.1,2,15 The ophthalmologist who treats an infant with retinoblastoma may eventually lose follow-up of the patient over the years after the eyes are successfully treated. The parents may be reluctant to inform the child that he or she had cancer during infancy and may even lead that child to believe that the eye was removed because of trauma or infection. The patient might grow up and have children without realizing the possibility existed of transmitting a malignant tumor to them.
When counseling the patient and family about the possibility of future children developing retinoblastoma, it is critical to know if the child or family carries a germline mutation for the retinoblastoma gene. Several clinical features help identify those families that might carry the retinoblastoma gene. Those patients with bilateral retinoblastoma and those patients with a family history of retinoblastoma can be assumed to have a germline mutation for the retinoblastoma gene. These children are at a 50% risk of passing this gene to future children. The retinoblastoma gene is about 80% penetrant so that only 40% of their offspring will manifest the clinical findings of the gene and some offspring may only be carriers of the gene without developing retinoblastoma.14
Based on genetic studies, the general percentages for predicting the chance of future children inheriting and developing retinoblastoma are listed in Tables 3-35.2 and 3-35.3. Table 3-35.2 refers to families with no history of retinblastoma, whereas Table 3-35.3 refers to families in which at least one family member had retinoblastoma.
Table 3-35.2. Risk for future offspring to develop retinoblastoma when there is a negative family history* | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||
Table 3-35.3. Risk for future offspring to develop retinoblastoma when there is a positive family history* | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||
Life-threatening Problems
Children with retinoblastoma are at risk for three important, life-threatening problems, including metastasis from retinoblastoma, intracranial neuroblastic malignancy (trilateral retinoblastoma), and second primary tumors.
At Risk for Metastases
Retinoblastoma metastasis, when it occurs, generally develops within 1 year of the diagnosis of the intraocular tumor. Those at greatest risk for metastasis show features of retinoblastoma invasion beyond the lamina cribrosa in the optic nerve, in the choroid (>2 mm dimension), sclera, orbit, or anterior chamber.19 Eyes with invasion of the optic nerve or choroid generally demonstrate large retinoblastoma (>15 mm greatest dimension) along with elevated intraocular pressure and total retinal detachment.20,21 Patients with evidence of invasive retinoblastoma should be treated with chemotherapy for 4 to 6 months to prevent metastases; however, criteria regarding the need for adjuvant chemotherapy remain unclear. In an analysis from our department, metastases were reduced from 24% in those without preventative chemotherapy to 4% in those who used chemotherapy.19
At Risk for Neuroblastic Intracranial Malignancy (Trilateral Retinoblastoma)
An association of neuroblastic intracranial malignancy exists in patients with the hereditary form of retinoblastoma, most often manifesting as pineoblastoma or other parasellar tumors.22 The pineoblastoma is identical to retinoblastoma from an embryologic and pathologic standpoint.23,24,25,26 This association of midline intracranial pineal tumors and suprasellar or parasellar neuroblastic tumors with bilateral retinoblastoma has been termed “trilateral” retinoblastoma.22 Function loss of the retinoblastoma gene is believed to confer an increased susceptibility to developing these intracranial tumors. Trilateral retinoblastoma is found in approximately 3% of all children with retinoblastoma.25,26 Those patients with bilateral or familial disease are at greatest risk, with 5% to 15% developing this finding.25,26 Hence, patients with bilateral or familial retinoblastoma are advised to have screening for pineoblastoma using computed tomography or magnetic resonance imaging (MRI) of the brain twice yearly for the first 5 years of life. In some cases, the intracranial tumor preceded the diagnosis of retinoblastoma.25 Possibly, many cases of pineoblastoma were previously misinterpreted as metastatic retinoblastoma to the brain. Unlike other second tumors, the pineoblastoma usually occurs during the first 5 years of life,26 whereas second tumors often take many decades to develop. Pineoblastoma is usually fatal, however. The possibility of pineoblastoma should be included in the genetic counseling of patients with hereditary retinoblastoma. Newer evidence suggests that recent treatment methods of systemic chemoreduction for retinoblastoma may prevent trilateral retinoblastoma.27,28 In a study of 100 patients with hereditary retinoblastoma, trilateral retinoblastoma was found in no patient who received chemoreduction and it would have been expected in 5 to 15 patients.28 Thus, prevention of trilateral retinoblastoma may be possible with neoadjuvant chemotherapy. Others believe that the avoidance of external beam radiotherapy is the main reason for reduction in the frequency of trilateral retinoblastoma.29,30 It should also be recognized that pineal gland enlargement might not be a solid tumor and it could represent a benign cyst.31
At Risk for Second Primary Tumors
Another important aspect of genetic counseling concerns the development of new genetically related cancers in survivors of bilateral or heritable retinoblastoma. It is now recognized that a child with retinoblastoma has an approximate 5% chance of developing another malignancy during the first 10 years of follow up, 18% during the first 20 years, and 26% within 30 years.32 The 30-year cumulative incidence is about 35% or even higher for those patients who received radiation therapy (external beam therapy) as compared with an incidence rate of 6% for those patients who avoided radiation. Therefore, patients with bilateral retinoblastoma have an increased incidence of second tumors, and this rate is further increased in those treated with external radiation therapy.32 Osteogenic sarcoma, often involving the femur, is most common; other tumors also recognized include spindle cell sarcoma, chondrosarcoma, rhabdomyosarcoma, neuroblastoma, glioma, leukemia, sebaceous cell carcinoma, squamous cell carcinoma, and malignant melanoma. Patients who survive a second tumor are at risk for a third, fourth, and even fifth nonocular tumor.33,34
Clinical Features of Retinoblastoma
The clinical manifestations of retinoblastoma vary with the stage of the disease at the time of recognition. In its earliest clinical stage, a small retinoblastoma (i.e., one <2 mm in basal dimension) appears ophthalmoscopically as a subtle, transparent or slightly translucent lesion in the sensory retina.1,2 Slightly larger tumors lead to dilated retinal blood vessels that feed and drain the tumor. Some larger tumors show foci of chalklike calcification that resemble cottage cheese. A retinoblastoma of any size can produce leukocoria (Fig. 3-35.2). The larger tumors more often present with leukocoria. This white pupillary reflex is a result of reflection of light from the white mass in the retrolental area. Some normal eyes show leukocoria from the reflection of the normal optic disc through the pupil.35
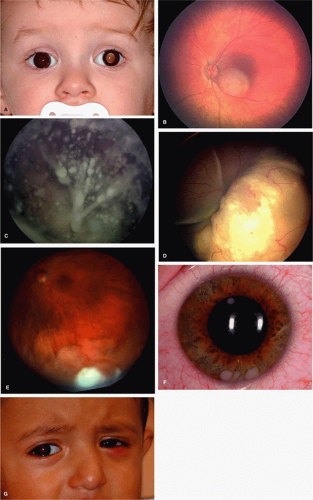 Figure 3-35.2. Clinical features of retinoblastoma. A: Leukocoria can be produced by a tumor of any size. This patient had a 3-mm retinoblastoma. B: Intraretinal retinoblastoma with tumor localized within the retina. C: Endophytic retinoblastoma with tumor seeding into vitreous cavity. D: Exophytic retinoblastoma with tumor growth beneath the retina and presence of subretinal fluid. E: Diffuse infiltrating retinoblastoma is considered a subset of endophytic retinoblastoma. It appears as flat infiltration of the retina. F: Diffuse infiltrating retinoblastoma with anterior chamber tumor seeds. G: Massive retinoblastoma with orbital inflammation caused by tumor necrosis. |
Retinoblastoma growth patterns are subdivided into intraretinal, endophytic, and exophytic (Fig. 3-35.2). Intraretinal tumors are those listed above, limited to the substance of the retina. Endophytic retinoblastoma is one that grows from the retina inward toward the vitreous cavity. Hence, it is characterized by a white hazy mass with obscuration of the retinal blood vessels. Because of its friable nature, an endophytic tumor can seed the vitreous cavity and anterior chamber and simulate endophthalmitis, especially toxocariasis, a parasitic disease found in young children. An exophytic retinoblastoma is one that grows from the retina outward into the subretinal space. Such tumors produce a progressive retinal detachment, with the retina often displaced anteriorly behind a clear lens. An exophytic retinoblastoma can clinically resemble Coats’ disease or other forms of exudative retinal detachment. Occasionally, a retinoblastoma can assume a diffuse infiltrating pattern, characterized by a relatively flat infiltration of the retina by tumor cells without an obvious mass. In such cases, the diagnosis may be more difficult and this pattern can simulate uveitis or endophthalmitis. Less frequently, the presenting feature can be pseudohypopyon caused by tumor seeding in the anterior chamber, hyphema secondary to iris neovascularization, vitreous hemorrhage, or signs of orbital cellulitis.
Classification of Retinoblastoma
Several classifications of retinoblastoma have been developed to assist in prediction of globe salvage and the most popular grouping is the Reese-Ellsworth classification36 (Table 3-35.4). A new International Classification of Retinoblastoma (ICRB) has been proposed to simplify the grouping scheme and allow a more practical approach to judging results of chemoreduction7,37,38,39 (Tables 3-35.5 and 3-35.6) (Fig. 3-35.3). Recent publication has shown that groups A, B, and C offer >90% control with chemoreduction and thermotherapy or cryotherapy alone, whereas group D has 47% control and group E should most often be enucleated39 (Fig. 3-35.4)
Table 3-35.4. Reese-Ellsworth Classification for Conservative Treatment of Retinoblastoma. | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||
Table 3-35.5. International Classification of Retinoblastoma (ICRB), long form | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


