6
Retinal Anomalies
BEST’S DISEASE
Barry N. Wasserman 
Etiology
 An autosomal dominant disorder also called vitelliform macular dystrophy, Best’s disease leads to retinal pigment epithelium (RPE) degeneration and secondary loss of photoreceptors in the macula.
An autosomal dominant disorder also called vitelliform macular dystrophy, Best’s disease leads to retinal pigment epithelium (RPE) degeneration and secondary loss of photoreceptors in the macula.
 Mutations lead to abnormal bestrophin, a Ca2+ sensitive Cl− channel protein. Lipofuscin accumulates in the RPE cells, yielding a characteristic “egg yolk” appearance (hence the name vitelliform) in the fovea and macula. Multifocal lesions may also occur.
Mutations lead to abnormal bestrophin, a Ca2+ sensitive Cl− channel protein. Lipofuscin accumulates in the RPE cells, yielding a characteristic “egg yolk” appearance (hence the name vitelliform) in the fovea and macula. Multifocal lesions may also occur.
Symptoms
 Early in the disease process, patients may be asymptomatic, but later metamorphopsia and deceased visual acuity occur.
Early in the disease process, patients may be asymptomatic, but later metamorphopsia and deceased visual acuity occur.
 Symptoms are variable and may be asymmetric, with significant vision loss in adulthood.
Symptoms are variable and may be asymmetric, with significant vision loss in adulthood.
Signs
 Stages are described based on phenotypic appearance in the macula, which may be asymmetric between the eyes. Earliest stage disease may only reveal subtle RPE mottling or may be normal. Later yellow orange material collects in the macula as a 0.5- to 5.0-mm diameter lesion yielding the classic “egg yolk” appearance. Clear fluid slowly builds around the lesion in the subretinal space, with resultant cystic appearance with fluid level (Fig. 6-1A). As this fluid dissipates, the lesion again changes to a more “scrambled egg”(vitelliruptive) stage, associated with pigment clumping (Fig. 6-1B). Later stages include atrophic changes with fibrosis, macular degeneration, and significant vision loss sometimes followed by choroidal neovascularization.
Stages are described based on phenotypic appearance in the macula, which may be asymmetric between the eyes. Earliest stage disease may only reveal subtle RPE mottling or may be normal. Later yellow orange material collects in the macula as a 0.5- to 5.0-mm diameter lesion yielding the classic “egg yolk” appearance. Clear fluid slowly builds around the lesion in the subretinal space, with resultant cystic appearance with fluid level (Fig. 6-1A). As this fluid dissipates, the lesion again changes to a more “scrambled egg”(vitelliruptive) stage, associated with pigment clumping (Fig. 6-1B). Later stages include atrophic changes with fibrosis, macular degeneration, and significant vision loss sometimes followed by choroidal neovascularization.
Differential Diagnosis
 Stargardt’s disease
Stargardt’s disease
 Sorsby’s macular dystrophy
Sorsby’s macular dystrophy
 Pattern dystrophy
Pattern dystrophy
 North Carolina macular dystrophy
North Carolina macular dystrophy
 Solar retinopathy
Solar retinopathy
 Coalescence of basal laminar drusen
Coalescence of basal laminar drusen
 Central serous retinopathy with fibrinous exudate
Central serous retinopathy with fibrinous exudate
 Pigment epithelial detachment of age-related macular degeneration
Pigment epithelial detachment of age-related macular degeneration
 Adult foveomacular dystrophy
Adult foveomacular dystrophy
 Age-related macular degeneration
Age-related macular degeneration
Diagnostic Evaluation
 Diagnosis is based on clinical findings along with electrophysiologic studies revealing normal electroretinogram and abnormal electro-oculogram. In addition, there is high autofluorescence.
Diagnosis is based on clinical findings along with electrophysiologic studies revealing normal electroretinogram and abnormal electro-oculogram. In addition, there is high autofluorescence.
 Optical coherence tomography (OCT) detects subretinal deposits and fluid.
Optical coherence tomography (OCT) detects subretinal deposits and fluid.
 Genetic studies may reveal mutation in the bestrophin gene.
Genetic studies may reveal mutation in the bestrophin gene.
Treatment
 There is no treatment for early disease, but late choroidal neovascularization has been treated with intravitreal injection of bevacizumab.
There is no treatment for early disease, but late choroidal neovascularization has been treated with intravitreal injection of bevacizumab.
 Genetic counseling and examination of family members are recommended, as are low vision and occupational consultations.
Genetic counseling and examination of family members are recommended, as are low vision and occupational consultations.
Prognosis
 Some patients maintain good vision (20/40) throughout life.
Some patients maintain good vision (20/40) throughout life.
 In others, good vision (20/20–20/50) usually persists through the early stages but decreases to 20/200 in the fifth and sixth decades of life as the atrophic and cicatricial stages progress.
In others, good vision (20/20–20/50) usually persists through the early stages but decreases to 20/200 in the fifth and sixth decades of life as the atrophic and cicatricial stages progress.
REFERENCES
Goodwin P. Hereditary retinal disease. Curr Opin Ophthalmol. 2008;19:255–262.
Leu J, Schrage NF, Degenring RF. Choroidal neovascularisation secondary to Best’s disease in a 13-year-old boy treated by intravitreal bevacizumab. Graefes Arch Clin Exp Ophthalmol. 2007;245(11):1723–1725.
Spaide RF, Noble K, Morgan A, et al. Vitelliform macular dystrophy. Ophthalmology. 2006;113:1392–1400.
FIGURE 6-1. A. Best’s disease with early macular “egg yolk” appearance. B. Best’s disease with later “scrambled egg” appearance.
CHOROIDEREMIA
Barry N. Wassermank 
Etiology
 An X-linked recessive disease, choroideremia is a progressive retinal degeneration. Males show loss of choriocapillaris and RPE. Female carriers can show mild signs of the disease, with patchy retinal abnormalities and corresponding visual field defects caused by lyonization, but are generally asymptomatic.
An X-linked recessive disease, choroideremia is a progressive retinal degeneration. Males show loss of choriocapillaris and RPE. Female carriers can show mild signs of the disease, with patchy retinal abnormalities and corresponding visual field defects caused by lyonization, but are generally asymptomatic.
 Mutation in the CHM gene affects production of the Rab escort protein 1 (REP-1). Degeneration begins in the midperiphery and progresses both centrally toward the macula and toward the periphery.
Mutation in the CHM gene affects production of the Rab escort protein 1 (REP-1). Degeneration begins in the midperiphery and progresses both centrally toward the macula and toward the periphery.
Symptoms
 Patients present in the first two decades of life with early loss of night vision and peripheral vision.
Patients present in the first two decades of life with early loss of night vision and peripheral vision.
 Central vision may be maintained until around the fifth decade of life but is eventually lost.
Central vision may be maintained until around the fifth decade of life but is eventually lost.
Signs
 Retinal examination reveals loss of the RPE and choriocapillaris with exposure of the larger choroidal vessels (Fig. 6-2). Remaining RPE may have a salt and pepper appearance.
Retinal examination reveals loss of the RPE and choriocapillaris with exposure of the larger choroidal vessels (Fig. 6-2). Remaining RPE may have a salt and pepper appearance.
 Later in the disease, large areas of exposed sclera may be seen on funduscopy. Posterior subcapsular cataracts may be associated.
Later in the disease, large areas of exposed sclera may be seen on funduscopy. Posterior subcapsular cataracts may be associated.
Differential Diagnosis
 Advanced retinitis pigmentosa
Advanced retinitis pigmentosa
 Gyrate atrophy
Gyrate atrophy
 Pathologic myopia
Pathologic myopia
 Chorioretinitis (e.g., acute retinal necrosis, after cytomegalovirus [CMV] retinitis)
Chorioretinitis (e.g., acute retinal necrosis, after cytomegalovirus [CMV] retinitis)
Diagnostic Evaluation
 X-linked inheritance with typical fundus appearance in affected males.
X-linked inheritance with typical fundus appearance in affected males.
 Visual field testing demonstrates constriction.
Visual field testing demonstrates constriction.
 Fluorescein angiography reveals large choroidal vessels caused by loss of overlying RPE and choriocapillaris.
Fluorescein angiography reveals large choroidal vessels caused by loss of overlying RPE and choriocapillaris.
 Electroretinography may be consistent with a rod-cone dystrophy early in the disease but eventually becomes extinguished.
Electroretinography may be consistent with a rod-cone dystrophy early in the disease but eventually becomes extinguished.
 Genetic analysis to assess mutation in the CHM gene may be performed, and analysis of peripheral blood is available to demonstrate absence of the Rab escort protein 1. Female carriers may demonstrate patches of irregular pigmentation in the RPE.
Genetic analysis to assess mutation in the CHM gene may be performed, and analysis of peripheral blood is available to demonstrate absence of the Rab escort protein 1. Female carriers may demonstrate patches of irregular pigmentation in the RPE.
Treatment
 Genetic counseling for family members should be suggested.
Genetic counseling for family members should be suggested.
 Low vision evaluation and treatment may be helpful.
Low vision evaluation and treatment may be helpful.
 Gene therapy has shown some success in animal models.
Gene therapy has shown some success in animal models.
Prognosis
 Patients progressively lose night and peripheral vision.
Patients progressively lose night and peripheral vision.
 Central vision is often maintained into adulthood but is ultimately lost.
Central vision is often maintained into adulthood but is ultimately lost.
REFERENCES
Lee TK, McTaggart KE, Sieving PA, et al. Clinical diagnoses that overlap with choroideremia. Can J Ophthalmol. 2003;38(5):364–372.
MacDonald IM, Russell L, Chan CC. Choroideremia: new findings from ocular pathology and review of recent literature. Surv Ophthalmol. 2009;54(3):401–407.
MacDonald IM, Smaoui N, Seabra MC. Choroideremia. In: Pagon RA, Bird TC, Dolan CR, et al, eds. GeneReviews. Seattle: University of Washington; 2010.
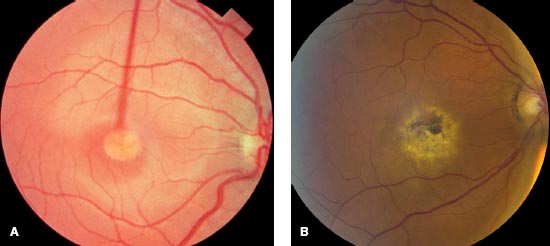
FIGURE 6-2. Choroideremia at advanced stage, with complete loss of retinal pigment epithelium and choriocapillaris.
GYRATE ATROPHY
Barry N. Wasserman 
Etiology
 Gyrate atrophy is an autosomal recessive disease caused by a deficiency of the mitochondrial enzyme, ornithine aminotransferase.
Gyrate atrophy is an autosomal recessive disease caused by a deficiency of the mitochondrial enzyme, ornithine aminotransferase.
 Elevated ornithine levels are toxic to the RPE, causing gradual loss of peripheral vision and night vision.
Elevated ornithine levels are toxic to the RPE, causing gradual loss of peripheral vision and night vision.
 Mutations of the ornithine aminotransferase gene (10q26) have been identified.
Mutations of the ornithine aminotransferase gene (10q26) have been identified.
Symptoms
 Nyctalopia and loss of visual field may begin in the first two decades of life but may not be manifest until the fifth decade. The disease affects both eyes symmetrically.
Nyctalopia and loss of visual field may begin in the first two decades of life but may not be manifest until the fifth decade. The disease affects both eyes symmetrically.
 Central vision is spared usually until the fourth or fifth decade but then declines.
Central vision is spared usually until the fourth or fifth decade but then declines.
 Symptoms and signs may vary widely in patients in all age groups.
Symptoms and signs may vary widely in patients in all age groups.
Signs
 Fundus examination early in the disease shows scalloped areas of geographic atrophic RPE and choriocapillaris (Fig. 6-3).
Fundus examination early in the disease shows scalloped areas of geographic atrophic RPE and choriocapillaris (Fig. 6-3).
 The macula is relatively spared until late in the disease, although epiretinal membranes and cystoid macular edema can occur.
The macula is relatively spared until late in the disease, although epiretinal membranes and cystoid macular edema can occur.
 Patients may have cataracts and myopia.
Patients may have cataracts and myopia.
Differential Diagnosis
 Choroideremia
Choroideremia
 Retinitis pigmentosa
Retinitis pigmentosa
 Choroidal atrophy
Choroidal atrophy
 Chorioretinitis (e.g., acute retinal necrosis, following CMV retinitis)
Chorioretinitis (e.g., acute retinal necrosis, following CMV retinitis)
Diagnostic Evaluation
 Ocular examination may reveal mildly decreased visual acuity, and refractive error is commonly myopic.
Ocular examination may reveal mildly decreased visual acuity, and refractive error is commonly myopic.
 Cataracts may be seen on slit-lamp evaluation.
Cataracts may be seen on slit-lamp evaluation.
 Funduscopy shows mid peripheral and peripheral scalloped geographic areas of RPE and choriocapillaris. Areas of intact RPE may have increased pigmentation.
Funduscopy shows mid peripheral and peripheral scalloped geographic areas of RPE and choriocapillaris. Areas of intact RPE may have increased pigmentation.
 The macula is usually spared early in the disease, but cystoid macular edema may be demonstrated with OCT.
The macula is usually spared early in the disease, but cystoid macular edema may be demonstrated with OCT.
 Optic atrophy and attenuated retinal vessels are seen later in the disease.
Optic atrophy and attenuated retinal vessels are seen later in the disease.
 Visual fields are markedly constricted, and electroretinography reveals absent photopic and scotopic responses.
Visual fields are markedly constricted, and electroretinography reveals absent photopic and scotopic responses.
 Plasma ornithine levels are markedly elevated.
Plasma ornithine levels are markedly elevated.
Treatment
 Genetic counseling is suggested.
Genetic counseling is suggested.
 A diet restricting arginine will lower the plasma ornithine and may slow the loss of visual function.
A diet restricting arginine will lower the plasma ornithine and may slow the loss of visual function.
 Low vision services should be recommended.
Low vision services should be recommended.
Prognosis
 Although dietary arginine restriction delays the loss of function, the macula is eventually effected in most patients.
Although dietary arginine restriction delays the loss of function, the macula is eventually effected in most patients.
REFERENCES
Kaiser-Kupfer MI, Caruso RC, Valle D, et al. Use of an arginine-restricted diet to slow progression of visual loss in patients with gyrate atrophy. Arch Ophthalmol. 2004;122:982–984.
Peltola KE, Nanto-Salonen K, Heinonen OJ, et al. Ophthalmologic heterogeneity in subjects with gyrate atrophy of choroid and retina harboring the L402P mutation of ornithine aminotransferase. Ophthalmology. 2001;108:721–729.
Tamara TL, Andrade RE, Muccioli C, et al. Cystoid macular edema in gyrate atrophy of the choroid and retina: a fluorescein angiography and optical coherence tomography evaluation. Am J Ophthalmol. 2005;140:147–149.
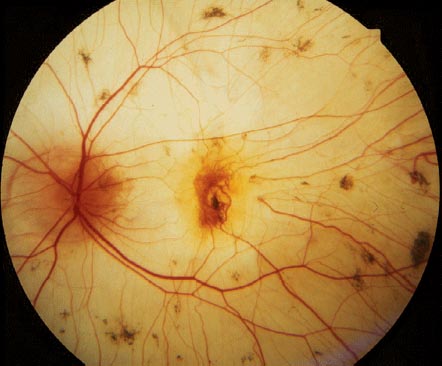
FIGURE 6-3. Gyrate atrophy with scalloped geographic atrophy of the retinal pigment epithelium and choriocapillaris. The macula is spared early in the disease.
LEBER CONGENITAL AMAUROSIS
Barry N. Wasserman 
Etiology
 Leber congenital amaurosis is a severe congenital retinal dystrophy affecting both cones and rods. Vision is often less than 20/200.
Leber congenital amaurosis is a severe congenital retinal dystrophy affecting both cones and rods. Vision is often less than 20/200.
 Mutations in more than 14 genes have been identified. The disease is almost autosomal recessive and rarely autosomal dominant.
Mutations in more than 14 genes have been identified. The disease is almost autosomal recessive and rarely autosomal dominant.
Symptoms and Signs
 Patients present with nystagmus at 2 to 3 months old. Children show poor visual behavior, and some show self-stimulatory eye poking (oculodigital reflex).
Patients present with nystagmus at 2 to 3 months old. Children show poor visual behavior, and some show self-stimulatory eye poking (oculodigital reflex).
 Pupils may be sluggish or paradoxical.
Pupils may be sluggish or paradoxical.
 Ophthalmoscopic findings are highly variable and may include a normal fundus appearance.
Ophthalmoscopic findings are highly variable and may include a normal fundus appearance.
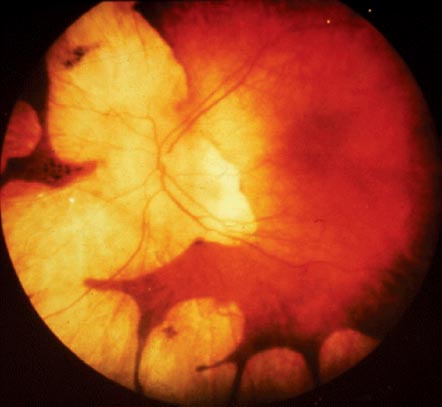
 Highly abnormal findings may also include chorioretinal atrophy, macular “coloboma” (Fig. 6-4A), retinal pigment epithelial irregularities, leopard spotting, nummular pigmentary abnormalities (Fig. 6-4B), and subretinal flecks.
Highly abnormal findings may also include chorioretinal atrophy, macular “coloboma” (Fig. 6-4A), retinal pigment epithelial irregularities, leopard spotting, nummular pigmentary abnormalities (Fig. 6-4B), and subretinal flecks.
 Retinal vessel attenuation and optic disc pallor are frequently present.
Retinal vessel attenuation and optic disc pallor are frequently present.
 Cataract, keratoconus, and strabismus may be associated findings.
Cataract, keratoconus, and strabismus may be associated findings.
Differential Diagnosis
 Achromatopsia
Achromatopsia
 Cone dystrophy
Cone dystrophy
 Goldman-Favre disease
Goldman-Favre disease
 Refsum phytanic acid storage disease
Refsum phytanic acid storage disease
 Bassen-Kornzweig (abetalipoproteinemia) syndrome
Bassen-Kornzweig (abetalipoproteinemia) syndrome
 Juvenile retinal dystrophy
Juvenile retinal dystrophy
 Toxoplasmosis (when macular “coloboma” present)
Toxoplasmosis (when macular “coloboma” present)
Diagnostic Evaluation
 Family history may reveal consanguinity or distantly related affected individuals.
Family history may reveal consanguinity or distantly related affected individuals.
 Examination, including retinal evaluation, combined with extinguished electroretinography findings is diagnostic.
Examination, including retinal evaluation, combined with extinguished electroretinography findings is diagnostic.
 Molecular genetic mutation screens are now available.
Molecular genetic mutation screens are now available.
 Systemic and blood testing are important to rule out treatable causes, such as Refsum phytanic acid storage disease.
Systemic and blood testing are important to rule out treatable causes, such as Refsum phytanic acid storage disease.
 Because there may be associated abnormalities of the kidneys, the liver, or hearing, appropriate testing is recommended.
Because there may be associated abnormalities of the kidneys, the liver, or hearing, appropriate testing is recommended.
Treatment
 Low vision evaluation and treatment can aid in function in some patients.
Low vision evaluation and treatment can aid in function in some patients.
 Gene transfer via subretinal administration of adeno-associated viral (AAV) vectors encoding has demonstrated safety and in some cases efficacy for patients with mutations in the gene RPE65.
Gene transfer via subretinal administration of adeno-associated viral (AAV) vectors encoding has demonstrated safety and in some cases efficacy for patients with mutations in the gene RPE65.
Prognosis
 Vision for most patients is poor.
Vision for most patients is poor.
 Future advances in gene therapy are promising.
Future advances in gene therapy are promising.
REFERENCES
Goodwin P. Hereditary retinal disease. Curr Opin Ophthalmol. 2008;19:255–262.
Simonelli F, Maguire AM, Testa F, et al. Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol Ther. 2010;18(3):643–650.
Traboulsi EI. The Marshall M. Parks memorial lecture: making sense of early-onset childhood retinal dystrophies—the clinical phenotype of Leber congenital amaurosis. Br J Ophthalmol. 2010;94(10):1281–1287.
FIGURE 6-4. Leber congenital amaurosis with macular “coloboma” (A) and nummular pigmentary abnormalities (B).
ASTROCYTIC HAMARTOMA
Anuradha Ganesh 
 Retinal astrocytic hamartoma is a glial tumor arising from the retinal nerve fiber layer. It is most frequently associated with tuberous sclerosis (Bourneville’s disease) but may very rarely be seen in neurofibromatosis or as an isolated ocular finding in otherwise normal individuals.
Retinal astrocytic hamartoma is a glial tumor arising from the retinal nerve fiber layer. It is most frequently associated with tuberous sclerosis (Bourneville’s disease) but may very rarely be seen in neurofibromatosis or as an isolated ocular finding in otherwise normal individuals.
 Astrocytic hamartomas are believed to be congenital in most cases but may not be recognized until later in childhood. Patients with these lesions are usually asymptomatic.
Astrocytic hamartomas are believed to be congenital in most cases but may not be recognized until later in childhood. Patients with these lesions are usually asymptomatic.
Epidemiology and Etiology
 Most astrocytic hamartomas occur congenitally in association with tuberous sclerosis, a phakomatosis characterized by the triad of seizures, mental retardation, and skin lesions (Table 6-1).
Most astrocytic hamartomas occur congenitally in association with tuberous sclerosis, a phakomatosis characterized by the triad of seizures, mental retardation, and skin lesions (Table 6-1).
 Tuberous sclerosis has an estimated incidence of 1 in 15,000 to 100,000 and exhibits autosomal dominant inheritance. About 60% of cases are spontaneous mutations. Mutations in either the TSC1 or TSC2 gene on chromosomes 9q34 and 16p13, respectively, result in tuberous sclerosis.
Tuberous sclerosis has an estimated incidence of 1 in 15,000 to 100,000 and exhibits autosomal dominant inheritance. About 60% of cases are spontaneous mutations. Mutations in either the TSC1 or TSC2 gene on chromosomes 9q34 and 16p13, respectively, result in tuberous sclerosis.
Table 6-1. Typical Manifestations of Tuberous Sclerosis

Signs
 About 50% of patients with tuberous sclerosis develop astrocytic hamartomas, approximately 50% of whom have bilateral involvement.
About 50% of patients with tuberous sclerosis develop astrocytic hamartomas, approximately 50% of whom have bilateral involvement.
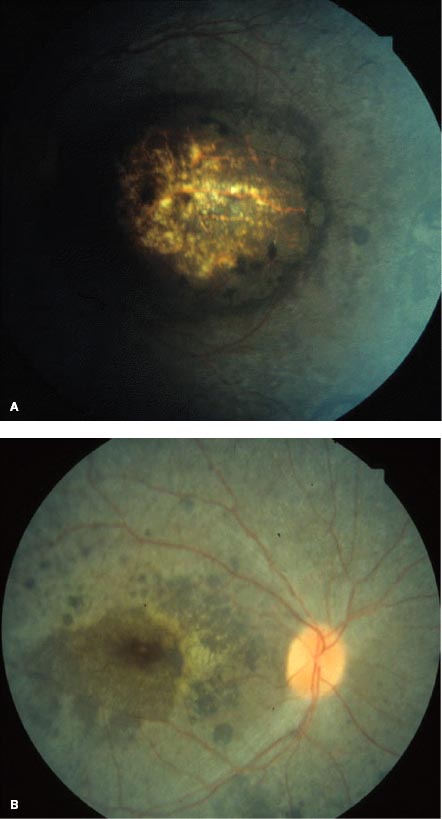
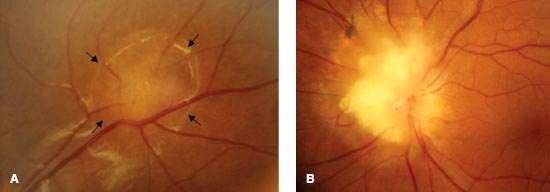
 Retinal astrocytic hamartomas are usually small, ranging from 0.5 to 5.0 mm in diameter, and are principally of two types: small, flat, smooth tumors that appear as subtle, ill-defined, semitranslucent thickening of the nerve fiber layer (Fig. 6-5A). Over time, they may become more opaque and contain calcification. Larger, opaque, calcified, sessile, whitish-yellow nodular masses (“mulberry lesion”) are found at the optic nerve (Fig. 6-5B). Visual field testing may reveal a scotoma in the area corresponding to the tumor.
Retinal astrocytic hamartomas are usually small, ranging from 0.5 to 5.0 mm in diameter, and are principally of two types: small, flat, smooth tumors that appear as subtle, ill-defined, semitranslucent thickening of the nerve fiber layer (Fig. 6-5A). Over time, they may become more opaque and contain calcification. Larger, opaque, calcified, sessile, whitish-yellow nodular masses (“mulberry lesion”) are found at the optic nerve (Fig. 6-5B). Visual field testing may reveal a scotoma in the area corresponding to the tumor.
 Occasionally, lesions may produce vitreous hemorrhage, vitreous seeding, subretinal hemorrhage, or retinal detachment. The presence of yellow, lipoproteinaceous exudation in the sensory retina and subretinal space is an uncommon feature.
Occasionally, lesions may produce vitreous hemorrhage, vitreous seeding, subretinal hemorrhage, or retinal detachment. The presence of yellow, lipoproteinaceous exudation in the sensory retina and subretinal space is an uncommon feature.
 Less frequent ocular manifestations include patches of iris or RPE hypopigmentation and hamartomas of the iris and ciliary body.
Less frequent ocular manifestations include patches of iris or RPE hypopigmentation and hamartomas of the iris and ciliary body.
 Patients with an astrocytic hamartoma of the retina or optic nerve should be evaluated for tuberous sclerosis, which is characterized by the triad of skin lesions, seizures, and mental retardation. Additional manifestations include hamartomas in various organ systems. Diagnostic criteria for tuberous sclerosis facilitate identifying definite, probable, and possible cases).
Patients with an astrocytic hamartoma of the retina or optic nerve should be evaluated for tuberous sclerosis, which is characterized by the triad of skin lesions, seizures, and mental retardation. Additional manifestations include hamartomas in various organ systems. Diagnostic criteria for tuberous sclerosis facilitate identifying definite, probable, and possible cases).
Differential Diagnosis
 Retinoblastoma
Retinoblastoma
 Retinocytoma
Retinocytoma
 Myelinated nerve fibers
Myelinated nerve fibers
 Retinal or optic nerve granuloma
Retinal or optic nerve granuloma
 Drusen of the optic nerve
Drusen of the optic nerve
 Papillitis or papilledema
Papillitis or papilledema
 Optic nerve infiltration (e.g., tuberculosis, sarcoid, leukemia)
Optic nerve infiltration (e.g., tuberculosis, sarcoid, leukemia)
Diagnostic Evaluation
 OCT: The astrocytic hamartoma replaces normal retinal architecture and appears as an optically hyperreflective mass with retinal disorganization, moth-eaten spaces, and posterior shadowing.
OCT: The astrocytic hamartoma replaces normal retinal architecture and appears as an optically hyperreflective mass with retinal disorganization, moth-eaten spaces, and posterior shadowing.
 Fluorescein angiography: The tumor appears relatively hypofluorescent in the arterial phase. Superficial find blood vessels are seen during the venous phase. The tumor stains intensely and homogeneously in the late phases. This test is rarely indicated.
Fluorescein angiography: The tumor appears relatively hypofluorescent in the arterial phase. Superficial find blood vessels are seen during the venous phase. The tumor stains intensely and homogeneously in the late phases. This test is rarely indicated.
 B-scan ultrasonography: A larger, calcified lesion often appears as a discrete, oval, solid mass with a sharp anterior border.
B-scan ultrasonography: A larger, calcified lesion often appears as a discrete, oval, solid mass with a sharp anterior border.
 Biopsy: In rare cases, differentiation from retinoblastoma (young patient) or choroidal melanoma (older patient) requires fine-needle aspiration biopsy
Biopsy: In rare cases, differentiation from retinoblastoma (young patient) or choroidal melanoma (older patient) requires fine-needle aspiration biopsy
 Imaging or ultrasonography: To detect astrocytomas in the brain and other parts of the body, including the chest and abdomen.
Imaging or ultrasonography: To detect astrocytomas in the brain and other parts of the body, including the chest and abdomen.
Treatment and Prognosis
 Most cases of retinal astrocytoma are asymptomatic and do not require treatment.
Most cases of retinal astrocytoma are asymptomatic and do not require treatment.
 Rarely, some tumors can exhibit aggressive behavior and cause hemorrhage; retinal detachment; or even more uncommonly, neovascular glaucoma.
Rarely, some tumors can exhibit aggressive behavior and cause hemorrhage; retinal detachment; or even more uncommonly, neovascular glaucoma.
 Serial follow-up is indicated.
Serial follow-up is indicated.
 Retinal detachment can be treated with demarcating laser photocoagulation.
Retinal detachment can be treated with demarcating laser photocoagulation.
 Progressive lesions may require measures such as endoresection; brachytherapy; and at times, enucleation.
Progressive lesions may require measures such as endoresection; brachytherapy; and at times, enucleation.
 Family members should be examined for manifestations of tuberous sclerosis. Genetic testing may be useful in confirming the diagnosis and identifying affected relatives, allowing monitoring for early detection of problems associated with tuberous sclerosis, thus leading to earlier treatment and better outcomes.
Family members should be examined for manifestations of tuberous sclerosis. Genetic testing may be useful in confirming the diagnosis and identifying affected relatives, allowing monitoring for early detection of problems associated with tuberous sclerosis, thus leading to earlier treatment and better outcomes.
REFERENCES
Northrup H, Sing Au K. Tuberous sclerosis complex Bourneville disease. In: Pagon RA, Bird TC, Dolan CR, et al, eds. GeneReviews. Seattle: University of Washington; 2009.
Roach ES, Sparagana SP. Diagnosis of tuberous sclerosis complex. J Child Neurol. 2004;19:643–649.
Shields CL, Benevides R, Materin MA, et al. Optical coherence tomography of retinal astrocytic hamartoma in 15 cases. Ophthalmology. 2006;113:1553–1557.
Shields JA, Eagle RC Jr, Shields CL, et al. Aggressive retinal astrocytomas in 4 patients with tuberous sclerosis complex, Arch Ophthalmol. 2005;123:856–863.
Shields JA, Shields CL. Atlas of Intraocular Tumors. Philadelphia: Lippincott Williams & Wilkins; 1999: 269–286.
FIGURE 6-5. Astrocytic hamartoma. A. Small, ill-defined thickening of the nerve fiber layer. The arrows indicate the edge of the lesion. B. Large, opaque, calcified, sessile, whitish-yellow nodular mass (“mulberry lesion”) at the optic nerve.
INCONTINENTIA PIGMENTI
Anuradha Ganesh and Alex V. Levin 
 Incontinentia pigmenti, or Bloch-Sulzberger syndrome, is a rare X-linked dominant disorder of skin pigmentation that is accompanied by ocular, dental and central nervous system manifestations.
Incontinentia pigmenti, or Bloch-Sulzberger syndrome, is a rare X-linked dominant disorder of skin pigmentation that is accompanied by ocular, dental and central nervous system manifestations.
Epidemiology and Etiology
 The incidence of incontinentia pigmenti is 1 in 40,000 live births.
The incidence of incontinentia pigmenti is 1 in 40,000 live births.
 In most cases, incontinentia pigmenti is caused by mutations in the NEMO (NF-κB essential modulator) gene at chromosome Xq28.
In most cases, incontinentia pigmenti is caused by mutations in the NEMO (NF-κB essential modulator) gene at chromosome Xq28.
 As an X-linked dominant condition, it is usually lethal in males and thus usually only seen in female infants. An affected male with Klinefelter’s syndrome (XXY) or genetic mosaicism may rarely survive.
As an X-linked dominant condition, it is usually lethal in males and thus usually only seen in female infants. An affected male with Klinefelter’s syndrome (XXY) or genetic mosaicism may rarely survive.
 Incontinentia pigmenti refers to the histologic feature of “incontinence” of melanocytes in the basal layer of the epidermis of their pigment, which is then found in the superficial dermis.
Incontinentia pigmenti refers to the histologic feature of “incontinence” of melanocytes in the basal layer of the epidermis of their pigment, which is then found in the superficial dermis.
History
 Characteristic skin lesions are usually present at birth.
Characteristic skin lesions are usually present at birth.
 Ophthalmic findings are noted in infancy or sometimes later in life.
Ophthalmic findings are noted in infancy or sometimes later in life.
 A pedigree may reveal a history of male fetus miscarriages.
A pedigree may reveal a history of male fetus miscarriages.
Signs
 The hallmark of ophthalmic involvement, present in more than 40% of patients, is peripheral retinal capillary nonperfusion (Fig. 6-6). This avascular retina may be demarcated by an intervening peripheral ring of neovascularization or otherwise abnormal retinal vessels, reminiscent of the clinical findings in retinopathy of prematurity (ROP). Retinal ischemia leads to the arteriovenous anastomoses and neovascularization. The disease may resolve spontaneously, but 10% of infants develop retinal folds, traction, and rhegmatogenous retinal detachment with consequent visual impairment. The ocular abnormalities are often asymmetric. Other eye findings are largely secondary and include strabismus in one third of infants, cataracts, and retinal pigmentary changes with mottled, diffuse hypopigmentation, and foveal hypoplasia.
The hallmark of ophthalmic involvement, present in more than 40% of patients, is peripheral retinal capillary nonperfusion (Fig. 6-6). This avascular retina may be demarcated by an intervening peripheral ring of neovascularization or otherwise abnormal retinal vessels, reminiscent of the clinical findings in retinopathy of prematurity (ROP). Retinal ischemia leads to the arteriovenous anastomoses and neovascularization. The disease may resolve spontaneously, but 10% of infants develop retinal folds, traction, and rhegmatogenous retinal detachment with consequent visual impairment. The ocular abnormalities are often asymmetric. Other eye findings are largely secondary and include strabismus in one third of infants, cataracts, and retinal pigmentary changes with mottled, diffuse hypopigmentation, and foveal hypoplasia.
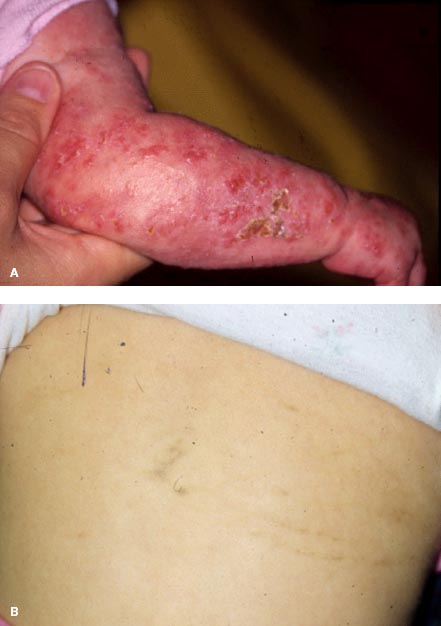
 Skin findings include vesicular eruptions at birth that later evolve into desquamating erythematous lesions (Fig. 6-7A) followed by swirling hyperpigmentation (marble cake appearance; Fig. 6-7B) and then scarring.
Skin findings include vesicular eruptions at birth that later evolve into desquamating erythematous lesions (Fig. 6-7A) followed by swirling hyperpigmentation (marble cake appearance; Fig. 6-7B) and then scarring.
 Dental abnormalities occur in more than 90% of affected individuals and include hypodontia; delayed eruption; and malformed, cone-shaped crowns. Associated central nervous system abnormalities include seizures, spastic paralysis, and mental retardation.
Dental abnormalities occur in more than 90% of affected individuals and include hypodontia; delayed eruption; and malformed, cone-shaped crowns. Associated central nervous system abnormalities include seizures, spastic paralysis, and mental retardation.
Differential Diagnosis
 ROP
ROP
 Familial exudative vitreoretinopathy (FEVR)
Familial exudative vitreoretinopathy (FEVR)
 Sickle cell disease (in older children)
Sickle cell disease (in older children)
 Other causes of infantile retinal nonattachment or detachment should also be considered.
Other causes of infantile retinal nonattachment or detachment should also be considered.
Diagnostic Evaluation
 The diagnosis of incontinentia pigmenti is based on clinical criteria (Table 6-2). Tests that may help in confirming the diagnosis include skin biopsy (although there is a wide differential diagnosis for the findings described above), neuroimaging (cerebral atrophy, agenesis of corpus callosum), fluorescein angiography (avascular peripheral retina, anomalous vessels in vascular-avascular junction), and genetic testing (for mutations in the NEMO gene).
The diagnosis of incontinentia pigmenti is based on clinical criteria (Table 6-2). Tests that may help in confirming the diagnosis include skin biopsy (although there is a wide differential diagnosis for the findings described above), neuroimaging (cerebral atrophy, agenesis of corpus callosum), fluorescein angiography (avascular peripheral retina, anomalous vessels in vascular-avascular junction), and genetic testing (for mutations in the NEMO gene).
TABLE 6-2. Diagnostic Criteria for Incontinentia Pigmenti*

 Careful skin, dental, and ocular examination of the patient’s mother may be useful as variable expression.
Careful skin, dental, and ocular examination of the patient’s mother may be useful as variable expression.
Treatment and Prognosis
 Patients with incontinentia pigmenti require careful retinal examination early in infancy to detect peripheral retinal nonperfusion. If present, frequent follow-up is indicated to enable early detection of change that might suggest development of retinal neovascularization, vascular leakage, and traction.
Patients with incontinentia pigmenti require careful retinal examination early in infancy to detect peripheral retinal nonperfusion. If present, frequent follow-up is indicated to enable early detection of change that might suggest development of retinal neovascularization, vascular leakage, and traction.
 No treatment is indicated unless such changes develop, in which case laser photocoagulation is performed in an effort to prevent progression to retinal traction and detachment.
No treatment is indicated unless such changes develop, in which case laser photocoagulation is performed in an effort to prevent progression to retinal traction and detachment.
 Genetic counseling is also highly recommended.
Genetic counseling is also highly recommended.
REFERENCES
Holmstrom G, Thoren K. Ocular manifestations of incontinentia pigmenti. Acta Ophthalmol Scand. 2000;78:348–353.
International IP Consortium. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. Nature. 2000;405:466–447.
Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet. 1993;30:53–59.
Scheuerle A, Ursini MV. Incontinentia pigmenti Bloch Sulzberger syndrome. In: Pagon RA, Bird TC, Dolan CR, et al, eds. GeneReviews. Seattle: University of Washington; 2005.
FIGURE 6-6. Fundus of a patient with incontinentia pigmenti showing peripheral retinal capillary nonperfusion (asterisk), arteriovenous anastomoses (arrows), and exudates (arrowhead).
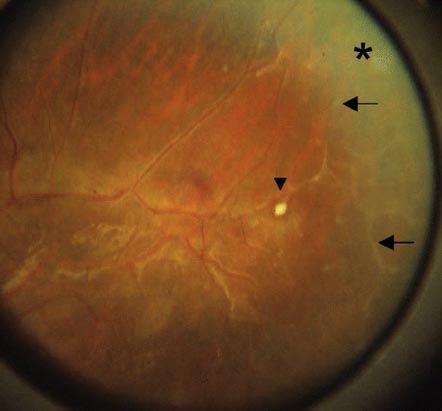
FIGURE 6-7. Skin findings in Coats’ disease. Vesicular and desquamating erythematous lesions in an infant with incontinentia pigmenti (A) and swirling hyperpigmentation (“marble cake” appearance) (B).
COATS’ DISEASE
Barry N. Wasserman and Carol L. Shields 
Coats’ disease is a nonhereditary condition characterized by unilateral retinal telangiectasia, exudation, and exudative retinal detachment that occurs most often in young patients.
Etiology
 Coats’ disease is more common in males and generally occurs as a unilateral trait.
Coats’ disease is more common in males and generally occurs as a unilateral trait.
 The average age at onset is 2 to 8 years.
The average age at onset is 2 to 8 years.
 Telangiectatic arterioles or venules lead to subretinal and intraretinal exudation of fluid and lipid, which eventual leads to exudative retinal detachment and vision loss.
Telangiectatic arterioles or venules lead to subretinal and intraretinal exudation of fluid and lipid, which eventual leads to exudative retinal detachment and vision loss.
 Some evidence indicates that Coats’ disease is related to a somatic mutation in the Norrie’s disease protein (NDP) gene or the frizzled 4 gene (FZD4).
Some evidence indicates that Coats’ disease is related to a somatic mutation in the Norrie’s disease protein (NDP) gene or the frizzled 4 gene (FZD4).
Symptoms
 The symptoms include painless vision loss, particularly when the disease occurs at an older age.
The symptoms include painless vision loss, particularly when the disease occurs at an older age.
 Younger patients often present with strabismus or xanthochroia.
Younger patients often present with strabismus or xanthochroia.
 Rarely, patients manifest neovascular glaucoma and a red, painful eye.
Rarely, patients manifest neovascular glaucoma and a red, painful eye.
 In an analysis of 150 cases of Coats’ disease, the symptoms included decreased visual acuity (43%), strabismus (23%), xanthochroia (20%), pain (3%), heterochromia (1%), nystagmus (1%), and no symptoms (8%).
In an analysis of 150 cases of Coats’ disease, the symptoms included decreased visual acuity (43%), strabismus (23%), xanthochroia (20%), pain (3%), heterochromia (1%), nystagmus (1%), and no symptoms (8%).
Signs
 The findings in Coats’ disease include telangiectasia (100%), retinal exudation (99%), exudative retinal detachment (81%), retina hemorrhage (13%), retinal macrocyst (11%), vasoproliferative tumor (6%), and neovascularization of disc (2%), retina (1%), and iris (8%). Anterior chamber cholesterolosis occurs in 3%.
The findings in Coats’ disease include telangiectasia (100%), retinal exudation (99%), exudative retinal detachment (81%), retina hemorrhage (13%), retinal macrocyst (11%), vasoproliferative tumor (6%), and neovascularization of disc (2%), retina (1%), and iris (8%). Anterior chamber cholesterolosis occurs in 3%.
Differential Diagnosis
 Retinoblastoma
Retinoblastoma
 ROP
ROP
 Retinal hemangioblastoma
Retinal hemangioblastoma
 Retinal vasoproliferative tumor
Retinal vasoproliferative tumor
 Sickle cell retinopathy
Sickle cell retinopathy
 FEVR
FEVR
 Toxocariasis
Toxocariasis
 Leukemia
Leukemia
 Persistent fetal vasculature (PFV; persistent hyperplastic primary vitreous [PHPV])
Persistent fetal vasculature (PFV; persistent hyperplastic primary vitreous [PHPV])
 Radiation retinopathy
Radiation retinopathy
 Retinopathy of hypertensive crisis
Retinopathy of hypertensive crisis
 Cataract
Cataract
 Coloboma
Coloboma
Diagnostic Evaluation
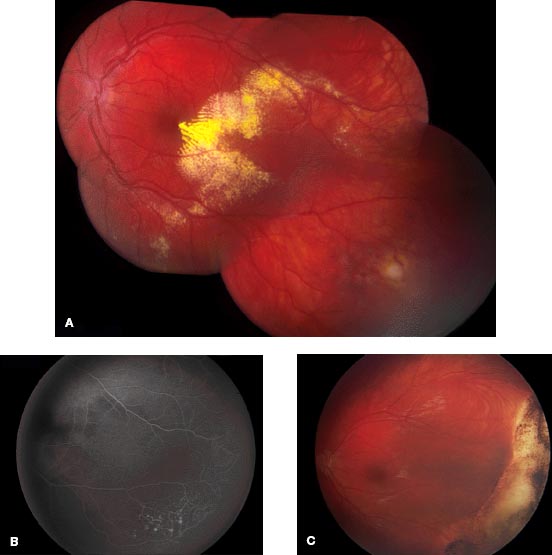
 The diagnosis is established by recognition of the clinical features on ophthalmoscopy. Retinal telangiectatic vessels with fusiform and nodular vascular dilation and adjacent nonperfusion are found, classically in the temporal quadrant. Related intraretinal and subretinal exudation is noted, and often retinal detachment is found (Figs. 6-8 and 6-9).
The diagnosis is established by recognition of the clinical features on ophthalmoscopy. Retinal telangiectatic vessels with fusiform and nodular vascular dilation and adjacent nonperfusion are found, classically in the temporal quadrant. Related intraretinal and subretinal exudation is noted, and often retinal detachment is found (Figs. 6-8 and 6-9).
 Fluorescein angiography confirms these findings and additionally reveals vascular leakage and macular edema. The classification of Coats’ disease, proposed by Shields et al, is:
Fluorescein angiography confirms these findings and additionally reveals vascular leakage and macular edema. The classification of Coats’ disease, proposed by Shields et al, is:
 Stage 1: retinal telangiectasia (T)
Stage 1: retinal telangiectasia (T)
 Stage 2: T + exudation (E)
Stage 2: T + exudation (E)
 Stage 3: T + E + subretinal fluid (F)
Stage 3: T + E + subretinal fluid (F)
 Stage 4: T + E + F + neovascular glaucoma (G)
Stage 4: T + E + F + neovascular glaucoma (G)
 Stage 5: T + E + F + G + phthisis bulbi
Stage 5: T + E + F + G + phthisis bulbi
Treatment
 Laser photocoagulation or cryotherapy is used to obliterate leaking vessels.
Laser photocoagulation or cryotherapy is used to obliterate leaking vessels.
 The role of intravitreal anti-VEGFs is being explored.
The role of intravitreal anti-VEGFs is being explored.
 Pars plana vitrectomy with drainage of subretinal fluid and exudation is used for advanced cases.
Pars plana vitrectomy with drainage of subretinal fluid and exudation is used for advanced cases.
Prognosis
 The visual prognosis is often poor. In one analysis of 150 cases, poor visual acuity of 20/200 or worse was found in no patients with stage 1 disease, but 50% of stage 2, 70% of stage 3, and 100% of stage 4 and 5 patients showed poor vision.
The visual prognosis is often poor. In one analysis of 150 cases, poor visual acuity of 20/200 or worse was found in no patients with stage 1 disease, but 50% of stage 2, 70% of stage 3, and 100% of stage 4 and 5 patients showed poor vision.
 The goal of treatment is stabilization of the eye with resolution of retinal detachment and exudation and prevention of neovascular glaucoma and loss of the eye.
The goal of treatment is stabilization of the eye with resolution of retinal detachment and exudation and prevention of neovascular glaucoma and loss of the eye.
 There are no systemic implications. However, patients with bilateral “Coats’” disease should be evaluated for systemic syndromes.
There are no systemic implications. However, patients with bilateral “Coats’” disease should be evaluated for systemic syndromes.
REFERENCES
Morris B, Foot B, Mulvihill A. A population-based study of Coats disease in the United Kingdom I: epidemiology and clinical features at diagnosis. Eye (Lond). 2010;24(12):1797–1801.
Mulvihill A, Morris B. A population-based study of Coats disease in the United Kingdom II: investigation, treatment, and outcomes. Eye (Lond). 2010; 24(12):1802–1807.
Shields JA, Shields CL, Honavar S, et al. Coats disease. Clinical variations and complications of Coats’ disease in 150 cases. The 2000 Sanford Gifford Memorial Lecture. Am J Ophthalmol. 2001;131:561–571.
Shields JA, Shields CL, Honavar SG, et al. Classification and management of Coats disease. The 2000 Proctor lecture. Am J Ophthalmol. 2001;131:572–583.
FIGURE 6-8. Coats’ disease (stage 2) with mild temporal macular exudation (A) from telangiectatic vessels (B) that showed resolution after therapy (C) with cryotherapy and laser photocoagulation, allowing for good visual acuity.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


