Retina
11.1 Posterior Vitreous Detachment
Symptoms
Floaters (“cobwebs,” “bugs,” or “spots” that change position with eye movement), blurred vision, flashes of light, which are more common in dim illumination and the temporal visual field.
Signs
Critical. One or more discrete light gray to black vitreous opacities, one often in the shape of a ring (“Weiss ring”) or broken ring, suspended over the optic disc (see Figure 11.1.1).
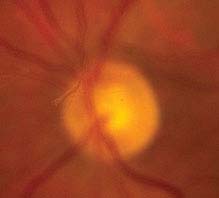
FIGURE 11.1.1. Posterior vitreous detachment.
Other. Retinal break, retinal detachment (RD), or vitreous hemorrhage (VH) may occur with or without a posterior vitreous detachment (PVD), with similar symptoms. Peripheral retinal and disc margin hemorrhages, released retinal pigment epithelial cells in the anterior vitreous (“tobacco dust” or Shafer sign).
NOTE: Approximately 8% to 10% of all patients with acute symptomatic PVD have a retinal break. The presence of pigmented cells in the anterior vitreous or VH in association with an acute PVD indicates a high probability (>70%) of a coexisting retinal break. See 11.2, Retinal Break.
Differential Diagnosis
 Vitritis: It may be difficult to distinguish PVD with anterior vitreous pigmented cells from inflammatory cells. In vitritis, vitreous cells may be found in both the posterior and anterior vitreous, the condition may be bilateral, and the cells are not typically pigmented. See 12.3, Posterior Uveitis.
Vitritis: It may be difficult to distinguish PVD with anterior vitreous pigmented cells from inflammatory cells. In vitritis, vitreous cells may be found in both the posterior and anterior vitreous, the condition may be bilateral, and the cells are not typically pigmented. See 12.3, Posterior Uveitis.
 Migraine: Multicolored photopsias in a zig-zag pattern that obstructs vision, lasts approximately 20 minutes. A headache may or may not follow. Normal fundus examination. See 10.27, Migraine.
Migraine: Multicolored photopsias in a zig-zag pattern that obstructs vision, lasts approximately 20 minutes. A headache may or may not follow. Normal fundus examination. See 10.27, Migraine.
Work-Up
1. History: Distinguish retinal photopsias from the visual distortion of migraine, which may be accompanied by new floaters. Duration of the symptoms? Risk factors for retinal break (previous intraocular surgery, high myopia, family history of retinal tears, and/or detachments)?
2. Complete ocular examination, including examination of the anterior vitreous for pigmented cells and a dilated retinal examination with indirect ophthalmoscopy and scleral depression to rule out a retinal break and detachment.
3. Visualize the PVD at the slit lamp with a 60- or 90-diopter lens by identifying a gray-to-black strand suspended in the vitreous. If not visible, have the patient look up, down, and then straight to float the PVD into view.
4. If a VH obscures visualization of the retina, ultrasonography (US) is indicated to identify the PVD and rule out a RD, tumor, or hemorrhagic macular degeneration. Occasionally, the flap of a tear can be identified. See 11.13, Vitreous Hemorrhage.
Treatment
No treatment is indicated for PVD. If an acute retinal break is found, see 11.2, Retinal Break.
NOTE: A retinal break surrounded by pigment is old and usually does not require treatment.
Follow-Up
 The patient should be given a list of RD symptoms (an increase in floaters or flashing lights, worsening vision, or the appearance of a persistent curtain or shadow anywhere in the field of vision) and told to return immediately if these symptoms develop.
The patient should be given a list of RD symptoms (an increase in floaters or flashing lights, worsening vision, or the appearance of a persistent curtain or shadow anywhere in the field of vision) and told to return immediately if these symptoms develop.
 If no retinal break or hemorrhage is found, the patient should be scheduled for repeated examination with scleral depression in 2 to 4 weeks, 2 to 3 months, and 6 months after the symptoms first develop.
If no retinal break or hemorrhage is found, the patient should be scheduled for repeated examination with scleral depression in 2 to 4 weeks, 2 to 3 months, and 6 months after the symptoms first develop.
 If no retinal break is found, but mild VH or peripheral punctate retinal hemorrhages are present, repeated examinations are performed 1 week, 2 to 4 weeks, 3 months, and 6 months after the event.
If no retinal break is found, but mild VH or peripheral punctate retinal hemorrhages are present, repeated examinations are performed 1 week, 2 to 4 weeks, 3 months, and 6 months after the event.
 If no retinal break is found but significant VH or anterior pigmented vitreous cells are present, repeat examination should be performed the next day by a retina specialist because of the high likelihood of a retinal break.
If no retinal break is found but significant VH or anterior pigmented vitreous cells are present, repeat examination should be performed the next day by a retina specialist because of the high likelihood of a retinal break.
11.2 RETINAL BREAK
Symptoms
Acute retinal break: Flashes of light, floaters (“cobwebs” or “spots” that change position with eye movement), and sometimes blurred vision. Can be identical to symptoms associated with PVD.
Chronic retinal breaks or atrophic retinal holes: Usually asymptomatic.
Signs
(See Figure 11.2.1.)
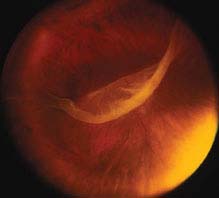
FIGURE 11.2.1. Giant retinal tear.
Critical. A full-thickness retinal defect, usually seen in the periphery.
Other. Acute retinal break: Pigmented cells in the anterior vitreous, VH, PVD, retinal flap, subretinal fluid (SRF), or an operculum (a free-floating piece of retina) suspended in the vitreous cavity above the retinal hole.
Chronic retinal break: A surrounding ring of pigmentation, a demarcation line between attached and detached retina, and signs (but no symptoms) of an acute retinal break.
Predisposing Conditions
Lattice degeneration, high myopia, aphakia, pseudophakia, age-related retinoschisis, vitreoretinal tufts, meridional folds, history of previous retinal break or detachment in the fellow eye, family history of retinal break or detachment, and trauma.
Differential Diagnosis
 Meridional fold: Small radial fold of retina perpendicular to the ora serrata and overlying an oral tooth; may have small retinal hole at the base.
Meridional fold: Small radial fold of retina perpendicular to the ora serrata and overlying an oral tooth; may have small retinal hole at the base.
 Meridional complex: Meridional fold that extends to a ciliary process.
Meridional complex: Meridional fold that extends to a ciliary process.
 Vitreoretinal tuft: Focal area of vitreous traction causing elevation of the retina.
Vitreoretinal tuft: Focal area of vitreous traction causing elevation of the retina.
 Paving stone degeneration.
Paving stone degeneration.
 Lattice degeneration.
Lattice degeneration.
Work-Up
Complete ocular examination with a slit-lamp and indirect ophthalmoscopy of both eyes with scleral depression. Scleral depression is deferred until 2 to 4 weeks after a traumatic hyphema or microhyphema.
Treatment
In general, laser therapy or cryotherapy is required within 24 to 72 hours for acute retinal breaks, and only rarely for chronic breaks. Each case must be individualized; however, we follow these general guidelines:
1. Treatment recommended
—Acute symptomatic break (e.g., a horseshoe or operculated tear).
—Acute traumatic break (including a dialysis).
2. Treatment to be considered
—Asymptomatic retinal break that is large (e.g., ≥1.5 mm), above the horizontal meridian, or both, particularly if there is no PVD.
—Asymptomatic retinal break in an aphakic or pseudophakic eye, an eye in which the involved or the contralateral eye has had an RD, or in a highly myopic eye.
Follow-Up
1. Patients with predisposing conditions or retinal breaks that do not require treatment are followed every 6 to 12 months.
2. Patients treated for a retinal break are reexamined in 1 week, 1 month, 3 months, and then every 6 to 12 months.
3. RD symptoms (an increase in floaters or flashing lights, the appearance of a curtain, shadow, worsening vision, or a bubble anywhere in the field of vision) are explained and patients are told to return immediately if these symptoms develop.
11.3 RETINAL DETACHMENT
There are three distinct types of RD. All three forms show an elevation of the retina.
RHEGMATOGENOUS RETINAL DETACHMENT
Symptoms
Flashes of light, floaters, a curtain, or shadow moving over the field of vision, peripheral or central visual loss, or both.
Signs
(See Figures 11.3.1 to 11.3.3.)
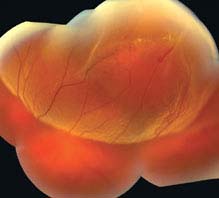
FIGURE 11.3.1. Rhegmatogenous retinal detachment.
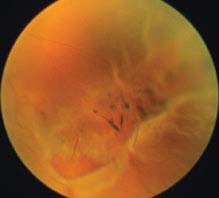
FIGURE 11.3.2. Retinal detachment with retinal break in lattice degeneration.
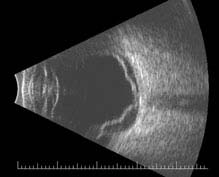
FIGURE 11.3.3. B-scan US of retinal detachment.
Critical. Elevation of the retina from the retinal pigment epithelium (RPE) by fluid in the subretinal space due to an accompanying full-thickness retinal break or breaks. See 11.2, Retinal Break.
Other. Anterior vitreous pigmented cells, VH, PVD, usually lower intraocular pressure (IOP) in the affected eye, nonshifting clear SRF, sometimes fixed retinal folds. The detached retina is often corrugated and partially opaque in appearance. A mild relative afferent pupillary defect (RAPD) may be present.
NOTE: A chronic rhegmatogenous retinal detachment (RRD) often shows a pigmented demarcation line at the posterior extent of the RD, intraretinal cysts, fixed folds, white dots underneath the retina (subretinal precipitates), or a combination of these with a relative visual field defect. It should be differentiated from retinoschisis, which produces an absolute visual field defect.
Etiology
A retinal break allows fluid to move through the hole and separate the overlying retina from the RPE.
Work-Up
1. Indirect ophthalmoscopy with scleral de-pression. Slit-lamp examination with contact lens may help in finding small breaks.
2. B-scan US may be helpful if media opacities are present.
EXUDATIVE RETINAL DETACHMENT
Symptoms
Minimal to severe visual loss or a visual field defect; visual changes may vary with changes in head position.
Signs
(See Figure 11.3.4.)
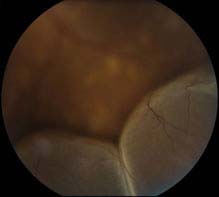
FIGURE 11.3.4. Exudative retinal detachment.
Critical. Serous elevation of the retina with shifting SRF. The area of detached retina changes when the patient changes position: While sitting, the SRF accumulates inferiorly, detaching the inferior retina; while in the supine position, the fluid accumulates in the posterior pole, detaching the macula. There is no retinal break; fluid accumulation is due to breakdown of the normal inner or outer blood-retinal barrier. The detachment does not extend to the ora serrata.
Other. The detached retina is smooth and may become quite bullous. A mild RAPD may be present if posterior pole involved.
Etiology
 Neoplastic: Choroidal malignant melanoma, metastasis, choroidal hemangioma, multiple myeloma, retinal capillary hemangioma (hemangioblastoma), etc.
Neoplastic: Choroidal malignant melanoma, metastasis, choroidal hemangioma, multiple myeloma, retinal capillary hemangioma (hemangioblastoma), etc.
 Inflammatory disease: Vogt–Koyanagi–Harada (VKH) syndrome, posterior scleritis, sympathetic ophthalmia, other chronic inflammatory processes.
Inflammatory disease: Vogt–Koyanagi–Harada (VKH) syndrome, posterior scleritis, sympathetic ophthalmia, other chronic inflammatory processes.
 Congenital abnormalities: Optic pit, morning-glory syndrome, and choroidal coloboma (although these may have an associated retinal break).
Congenital abnormalities: Optic pit, morning-glory syndrome, and choroidal coloboma (although these may have an associated retinal break).
 Vascular: Choroidal neovascularization (CNV), Coats disease, malignant hypertension (HTN), preeclampsia, and familial exudative vitreoretinopathy (FEVR). See specific sections.
Vascular: Choroidal neovascularization (CNV), Coats disease, malignant hypertension (HTN), preeclampsia, and familial exudative vitreoretinopathy (FEVR). See specific sections.
 Nanophthalmos: Small eyes with a small cornea and a shallow anterior chamber but a large lens and a thick sclera.
Nanophthalmos: Small eyes with a small cornea and a shallow anterior chamber but a large lens and a thick sclera.
 Idiopathic central serous chorioretinopathy (CSCR): May be seen with bullous RD from multiple, large RPE detachments. See 11.15, Central Serous Chorioretinopathy.
Idiopathic central serous chorioretinopathy (CSCR): May be seen with bullous RD from multiple, large RPE detachments. See 11.15, Central Serous Chorioretinopathy.
 Uveal effusion syndrome: Bilateral detachments of the peripheral choroid, ciliary body, and retina; leopard-spot RPE changes (when retina is reattached); cells in the vitreous; dilated episcleral vessels; more common in patients with high hyperopia, particularly nanophthalmic eyes.
Uveal effusion syndrome: Bilateral detachments of the peripheral choroid, ciliary body, and retina; leopard-spot RPE changes (when retina is reattached); cells in the vitreous; dilated episcleral vessels; more common in patients with high hyperopia, particularly nanophthalmic eyes.
Work-Up
1. Intravenous fluorescein angiography (IVFA) may show source of SRF.
2. Optical coherence tomography (OCT) may help identify SRF as well as the source (e.g., CNV).
3. B-scan US may help delineate the underlying cause.
4. Systemic work-up to rule out the above causes (e.g., HTN, multiple myeloma, etc.).
TRACTIONAL RETINAL DETACHMENT
Symptoms
Visual loss or visual field defect; may be asymptomatic.
Signs
(See Figure 11.3.5.)
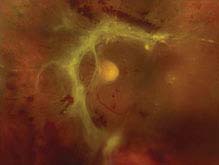
FIGURE 11.3.5. Traction retinal detachment.
Critical. The detached retina appears concave with a smooth surface; cellular and vitreous membranes exerting traction on the retina are present; retinal striae extending from these areas may also be seen. Detachment may become a convex RRD if a tractional retinal tear develops.
Other. The retina is immobile, and the detachment rarely extends to the ora serrata. A mild RAPD may be present.
Etiology
Fibrocellular bands in the vitreous (e.g., resulting from proliferative diabetic retinopathy (PDR), sickle cell retinopathy, retinopathy of prematurity, FEVR, toxocariasis, trauma, previous giant retinal tear) contract and detach the retina.
Work-Up
1. Indirect ophthalmoscopy with scleral depression. Slit-lamp examination with contact lens may help in finding small breaks.
2. B-scan US may be helpful if media opacities are present.
3. OCT is useful in identifying tractional membranes and can be useful in differentiating tractional membranes from detached retina.
Differential Diagnosis for All Three Types of Retinal Detachment
 Acquired/age-related degenerative retinoschisis: Commonly bilateral, usually inferotemporal, no pigmented cells or hemorrhage are present in the vitreous, the retinal vessels in the inner retinal layers are often sheathed peripherally, and white “snowflakes” are often seen on the inner retinal layers. See 11.4, Retinoschisis.
Acquired/age-related degenerative retinoschisis: Commonly bilateral, usually inferotemporal, no pigmented cells or hemorrhage are present in the vitreous, the retinal vessels in the inner retinal layers are often sheathed peripherally, and white “snowflakes” are often seen on the inner retinal layers. See 11.4, Retinoschisis.
 X-linked retinoschisis: Petaloid foveal changes are present over 90% of the time. Dehiscences occur in the nerve fiber layer (NFL) 50% of the time. See 11.4, Retinoschisis.
X-linked retinoschisis: Petaloid foveal changes are present over 90% of the time. Dehiscences occur in the nerve fiber layer (NFL) 50% of the time. See 11.4, Retinoschisis.
 Choroidal detachment: Orange–brown, more solid in appearance than an RD, often extends 360 degrees. Hypotony is usually present. See 11.27, Choroidal Effusion/Detachment.
Choroidal detachment: Orange–brown, more solid in appearance than an RD, often extends 360 degrees. Hypotony is usually present. See 11.27, Choroidal Effusion/Detachment.
Treatment
1. Patients with an acute RRD that threatens the fovea should be placed on bed rest, with surgical repair performed urgently. The visual prognosis is significantly worse in detachments that progress to involve the fovea. Surgical options include laser photocoagulation, cryotherapy, pneumatic retinopexy, vitrectomy, and scleral buckle.
2. All RRDs that are macula-off, or tractional retinal detachments that involve the macula, are repaired preferably within a few days. Visual outcomes for macula-off detachments do not change if surgery is performed within 7–10 days of the onset.
3. Chronic RDs are treated within 1 week if possible.
4. For exudative RD, successful treatment of the underlying condition often leads to resolution of the detachment.
Follow-Up
Patients treated for RD are reexamined at 1 day, 1 week, 2 weeks, 1 month, 2 to 3 months, then every 6 to 12 months.
11.4 RETINOSCHISIS
Retinoschisis, a splitting of the retina, occurs in X-linked (juvenile) and age-related degenerative forms.
X-LINKED (JUVENILE) RETINOSCHISIS
Symptoms
Decreased vision due to VH (25%) and macular changes, or asymptomatic. The condition is congenital, but may not be detected at birth if an examination is not performed. A family history may or may not be elicited (X-linked recessive).
Signs
(See Figure 11.4.1.)
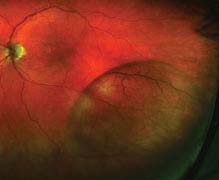
FIGURE 11.4.1. Retinoschisis.
Critical. Foveal schisis seen as stellate maculopathy: Cystoid foveal changes with retinal folds that radiate from the center of the fovea (petaloid pattern). Unlike the cysts of cystoid macular edema (CME), they do not stain or leak on IVFA, but can be seen with indocyanine green (ICG) and on OCT. The macular appearance changes in adulthood and the petaloid pattern may disappear.
Other. Separation of the nerve fiber layer (NFL) from the outer retinal layers in the retinal periphery (bilaterally in the inferotemporal quadrant, most commonly) with the development of NFL breaks; this peripheral retinoschisis occurs in 50% of patients. However, schisis may occur between any two retinal layers. RD, VH, and pigmentary changes also may occur. Pigmented demarcation lines may be seen (indicating previous RD) even though the retina is not detached at the time, unlike acquired age-related degenerative retinoschisis.
Differential Diagnosis
 Age-related degenerative retinoschisis (see below).
Age-related degenerative retinoschisis (see below).
 RRD: Usually unilateral, acquired, and associated with a retinal tear. Pigment in the anterior vitreous is seen. See 11.3, Retinal Detachment.
RRD: Usually unilateral, acquired, and associated with a retinal tear. Pigment in the anterior vitreous is seen. See 11.3, Retinal Detachment.
Work-Up
1. Family history.
2. Dilated retinal examination with scleral depression to rule out a retinal break or detachment.
3. OCT can help determine the layer of the schisis and help to differentiate schisis from an RD.
Treatment
1. No treatment for stellate maculopathy.
2. For non-clearing VH, consider vitrectomy.
3. Surgical repair of an RD should be performed.
4. Superimposed amblyopia may be present in children younger than 11 years of age when one eye is more severely affected and a trial of patching should be considered. See 8.7, Amblyopia.
Follow-Up
Every 6 months; sooner if treating amblyopia.
AGE-RELATED DEGENERATIVE
RETINOSCHISIS
Symptoms
Usually asymptomatic; may have decreased vision.
Signs
Critical. The schisis cavity is dome-shaped with a smooth surface and is usually located temporally, especially inferotemporally. Usually bilateral and may show sheathing of retinal vessels and “snowflakes” or “frosting” (persistent Mueller fibers) on the elevated inner wall of the schisis cavity. Unlike X-linked juvenile retinoschisis, splitting usually occurs at the level of the outer plexiform layer. The area of schisis is not mobile, and there is no associated RPE pigmentation.
Other. Prominent cystoid degeneration near the ora serrata, an absolute scotoma corresponding to the area of schisis, hyperopia is common, no pigment cells or hemorrhage in the vitreous, and absence of a demarcation line. An RRD may occasionally develop.
Differential Diagnosis
 RRD: Surface is corrugated in appearance and moves more with eye movements. A long-standing RD may resemble retinoschisis, but intraretinal cysts, demarcation lines between attached and detached retina, and white sub-retinal dots may be seen. Only a relative scotoma is present. See 11.3, Retinal Detachment.
RRD: Surface is corrugated in appearance and moves more with eye movements. A long-standing RD may resemble retinoschisis, but intraretinal cysts, demarcation lines between attached and detached retina, and white sub-retinal dots may be seen. Only a relative scotoma is present. See 11.3, Retinal Detachment.
 X-linked juvenile retinoschisis (see above).
X-linked juvenile retinoschisis (see above).
Work-Up
1. Slit-lamp evaluation for anterior chamber inflammation and pigmented anterior vitreous cells; neither should be present in isolated retinoschisis.
2. Dilated retinal examination with scleral depression to rule out a concomitant RD or an outer layer retinal hole, which may lead to an RD.
3. A fundus contact lens evaluation of the retina as needed to aid in recognizing outer layer retinal breaks.
4. OCT can help determine which layer of the retina is split.
5. Visual field testing will reveal an absolute scotoma in the area of schisis.
Treatment
1. Surgery is indicated when a clinically significant RD develops.
2. A small RD walled off by a demarcation line is usually not treated. This may take the form of pigmentation at the posterior border of outer layer breaks.
Follow-Up
Every 6 months. RD symptoms (an increase in floaters or flashing lights, blurry vision, or the appearance of a curtain or shadow anywhere in the field of vision) are explained to all patients, and patients are told to return immediately if these symptoms develop.
11.5 COTTON–WOOL SPOT
Symptoms
Visual acuity usually normal. Often asymptomatic.
Signs
(See Figure 11.5.1.)
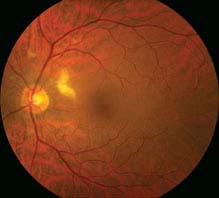
FIGURE 11.5.1. Cotton–wool spot.
Critical. Whitening in the superficial retinal NFL.
NOTE: The presence of even a single cotton–wool spot (CWS) is not normal. In a patient without diabetes mellitus, acute HTN, or a retinal vein occlusion, a work-up for an underlying systemic condition should be performed.
Differential Diagnosis
 Retinal whitening secondary to infectious retinitis, such as that seen in toxoplasmosis, herpes simplex virus, varicella zoster virus, and cytomegalovirus. These entities typically have vitritis and retinal hemorrhages associated with them. See 12.5, Toxoplasmosis, and 12.8, Acute Retinal Necrosis.
Retinal whitening secondary to infectious retinitis, such as that seen in toxoplasmosis, herpes simplex virus, varicella zoster virus, and cytomegalovirus. These entities typically have vitritis and retinal hemorrhages associated with them. See 12.5, Toxoplasmosis, and 12.8, Acute Retinal Necrosis.
 Myelinated NFL: Develops postnatally. Usually peripapillary but may be in retinal areas remote from the disc (see Figure 11.5.2).
Myelinated NFL: Develops postnatally. Usually peripapillary but may be in retinal areas remote from the disc (see Figure 11.5.2).
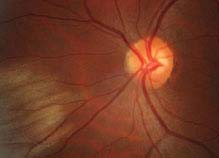
FIGURE 11.5.2. Myelinated nerve fiber layer.
Etiology
Thought to be an acute obstruction of a precapillary retinal arteriole causing blockage of axoplasmic flow and buildup of axoplasmic debris in the NFL.
 Diabetes mellitus: Most common cause. Often associated with microaneurysms, dot-blot hemorrhages, and hard exudates. See 11.12, Diabetic Retinopathy.
Diabetes mellitus: Most common cause. Often associated with microaneurysms, dot-blot hemorrhages, and hard exudates. See 11.12, Diabetic Retinopathy.
 Chronic or acute HTN: May see retinal arteriolar narrowing and flame hemorrhages in chronic HTN. Acute HTN may have hard exudates, optic nerve swelling, exudative RD. See 11.10, Hypertensive Retinopathy.
Chronic or acute HTN: May see retinal arteriolar narrowing and flame hemorrhages in chronic HTN. Acute HTN may have hard exudates, optic nerve swelling, exudative RD. See 11.10, Hypertensive Retinopathy.
 Retinal vein occlusion: Unilateral, multiple hemorrhages, venous dilation, and tortuosity. Multiple CWS, usually ≥6, seen in ischemic varieties. See 11.8, Central Retinal Vein Occlusion, and 11.9, Branch Retinal Vein Occlusion.
Retinal vein occlusion: Unilateral, multiple hemorrhages, venous dilation, and tortuosity. Multiple CWS, usually ≥6, seen in ischemic varieties. See 11.8, Central Retinal Vein Occlusion, and 11.9, Branch Retinal Vein Occlusion.
 Retinal emboli: Often from carotid arteries or heart with resulting ischemia and subsequent CWS distal to arterial occlusion. Patients require carotid Doppler examination and echocardiography. See 10.22, Transient Visual Loss/Amaurosis Fugax.
Retinal emboli: Often from carotid arteries or heart with resulting ischemia and subsequent CWS distal to arterial occlusion. Patients require carotid Doppler examination and echocardiography. See 10.22, Transient Visual Loss/Amaurosis Fugax.
 Collagen vascular disease: Systemic lupus erythematosus (most common), Wegener granulomatosis, polyarteritis nodosa, scleroderma, etc.
Collagen vascular disease: Systemic lupus erythematosus (most common), Wegener granulomatosis, polyarteritis nodosa, scleroderma, etc.
 Giant cell arteritis (GCA): Age ≥55 years. Symptoms include vision loss, scalp tenderness, jaw claudication, proximal muscle aches, etc. See 10.17, Arteritic Ischemic Optic Neuropathy (Giant Cell Arteritis).
Giant cell arteritis (GCA): Age ≥55 years. Symptoms include vision loss, scalp tenderness, jaw claudication, proximal muscle aches, etc. See 10.17, Arteritic Ischemic Optic Neuropathy (Giant Cell Arteritis).
 HIV retinopathy: Single or multiple cotton-wool spots in the posterior pole. See 12.10, Noninfectious Retinal Microvasculopathy/HIV Retinopathy.
HIV retinopathy: Single or multiple cotton-wool spots in the posterior pole. See 12.10, Noninfectious Retinal Microvasculopathy/HIV Retinopathy.
 Other infections: Toxoplasmosis, orbital mucormycosis, Lyme, leptospirosis, Rocky Mountain spotted fever, onchocerciasis, subacute bacterial endocarditis.
Other infections: Toxoplasmosis, orbital mucormycosis, Lyme, leptospirosis, Rocky Mountain spotted fever, onchocerciasis, subacute bacterial endocarditis.
 Hypercoagulable state: Polycythemia, multiple myeloma, cryoglobulinemia, Waldenström macroglobulinemia, antiphospholipid syndrome, factor V Leiden, activated protein C resistance, hyperhomocysteinemia, protein C and S deficiency, antithrombin III mutation, prothrombin mutation G20210A, etc.
Hypercoagulable state: Polycythemia, multiple myeloma, cryoglobulinemia, Waldenström macroglobulinemia, antiphospholipid syndrome, factor V Leiden, activated protein C resistance, hyperhomocysteinemia, protein C and S deficiency, antithrombin III mutation, prothrombin mutation G20210A, etc.
 Radiation retinopathy: Follows radiation therapy to the eye or periocular structures when the eye is irradiated inadvertently. May occur any time after radiation, but occurs most commonly within a few years. Maintain a high suspicion even in patients in whom the eye was reportedly shielded. Usually, 3,000 cGy is necessary, but it has been noted to occur with 1,500 cGy. Resembles diabetic retinopathy.
Radiation retinopathy: Follows radiation therapy to the eye or periocular structures when the eye is irradiated inadvertently. May occur any time after radiation, but occurs most commonly within a few years. Maintain a high suspicion even in patients in whom the eye was reportedly shielded. Usually, 3,000 cGy is necessary, but it has been noted to occur with 1,500 cGy. Resembles diabetic retinopathy.
 Interferon therapy.
Interferon therapy.
 Purtscher and pseudo-Purtscher retinopathy: Multiple cotton-wool spots and/or superficial hemorrhages in a peripapillary configuration. Typically bilateral but can be unilateral and asymmetric. See 3.19, Purtscher Retinopathy.
Purtscher and pseudo-Purtscher retinopathy: Multiple cotton-wool spots and/or superficial hemorrhages in a peripapillary configuration. Typically bilateral but can be unilateral and asymmetric. See 3.19, Purtscher Retinopathy.
 Cancer: Metastatic carcinoma, leukemia, lymphoma.
Cancer: Metastatic carcinoma, leukemia, lymphoma.
 Others: Migraine, hypotension, intravenous drug use, papilledema, papillitis, severe anemia, sickle cell, acute blood loss, etc.
Others: Migraine, hypotension, intravenous drug use, papilledema, papillitis, severe anemia, sickle cell, acute blood loss, etc.
Work-Up
1. History: Diabetes or HTN? Ocular or periocular radiation in past? GCA symptoms in appropriate age group? Symptoms of collagen vascular disease including joint pain, rashes, etc.? HIV risk factors?
2. Complete ocular examination, including dilated retinal examination with a slit lamp and a 60- or 90-diopter lens and indirect ophthalmoscopy. Look for concurrent hemorrhages, vascular occlusion, vasculitis, hard exudates.
3. Check a fasting blood sugar.
4. Check the blood pressure.
5. Consider erythrocyte sedimentation rate (ESR), c-reactive protein (CRP), and platelets if GCA is suspected.
6. Consider blood and urine cultures, chest x-ray, carotid Doppler examination, and echocardiography if emboli are suspected.
7. Consider HIV testing.
8. Fluorescein angiography is generally not helpful for an isolated CWS. IVFA reveals areas of capillary nonperfusion adjacent to the location of the CWS.
Treatment
Treat underlying etiology.
Follow-Up
Depends on the underlying etiology. CWS typically fade in 5 to 7 weeks but can remain longer if associated with diabetic retinopathy.
11.6 CENTRAL RETINAL ARTERY OCCLUSION
Symptoms
Unilateral, painless, acute vision loss (counting fingers to light perception in 94% of eyes) occurring over seconds; may have a history of transient visual loss (amaurosis fugax).
Signs
(See Figure 11.6.1.)
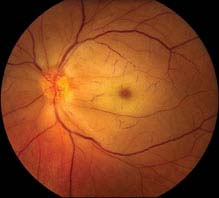
FIGURE 11.6.1. Central retinal artery occlusion.
Critical. Superficial opacification or whitening of the retina in the posterior pole, and a cherry-red spot in the center of the macula (may be subtle).
Other. Marked RAPD; narrowed retinal arterioles; boxcarring or segmentation of the blood column in the arterioles. Occasionally, retinal arteriolar emboli or cilioretinal artery sparing of the foveola is evident. If visual acuity is light perception or worse, strongly suspect ophthalmic artery occlusion.
Differential Diagnosis
 Acute ophthalmic artery occlusion: Usually no cherry-red spot; the entire retina appears whitened. The treatment is the same as for central retinal artery occlusion (CRAO).
Acute ophthalmic artery occlusion: Usually no cherry-red spot; the entire retina appears whitened. The treatment is the same as for central retinal artery occlusion (CRAO).
 Commotio retinae: Retinal whitening from intracellular edema and fragmentation of the photoreceptor outer segments and RPE. Follows blunt trauma, resolves spontaneously. May mimic a cherry-red spot when the posterior pole is involved. See 3.16, Commotio Retinae.
Commotio retinae: Retinal whitening from intracellular edema and fragmentation of the photoreceptor outer segments and RPE. Follows blunt trauma, resolves spontaneously. May mimic a cherry-red spot when the posterior pole is involved. See 3.16, Commotio Retinae.
 Other causes of a cherry-red spot: Tay–Sachs, Niemann–Pick disease type A, etc. These conditions present early in life with other, often severe, systemic manifestations. Ophthalmic findings are usually bilateral.
Other causes of a cherry-red spot: Tay–Sachs, Niemann–Pick disease type A, etc. These conditions present early in life with other, often severe, systemic manifestations. Ophthalmic findings are usually bilateral.
Etiology
 Embolus: Three main types include cholesterol, calcium, and platelet-fibrin emboli. All are seen within a vessel. Cholesterol emboli (Hollenhorst plaque) are typically refractile, orange, and seen at retinal vessel bifurcations. They arise from ulcerated atheromas, typically from the carotid arteries. Calcium emboli are white and frequently cause distal retinal infarction. They typically arise from cardiac valves. Platelet-fibrin emboli are a dull white and typically arise from atheromas in the carotid arteries.
Embolus: Three main types include cholesterol, calcium, and platelet-fibrin emboli. All are seen within a vessel. Cholesterol emboli (Hollenhorst plaque) are typically refractile, orange, and seen at retinal vessel bifurcations. They arise from ulcerated atheromas, typically from the carotid arteries. Calcium emboli are white and frequently cause distal retinal infarction. They typically arise from cardiac valves. Platelet-fibrin emboli are a dull white and typically arise from atheromas in the carotid arteries.
 Thrombosis.
Thrombosis.
 GCA: May produce CRAO, ophthalmic artery occlusion, or an ischemic optic neuropathy. See 10.17, Arteritic Ischemic Optic Neuropathy (Giant Cell Arteritis).
GCA: May produce CRAO, ophthalmic artery occlusion, or an ischemic optic neuropathy. See 10.17, Arteritic Ischemic Optic Neuropathy (Giant Cell Arteritis).
 Other collagen–vascular disease: Systemic lupus erythematosus, polyarteritis nodosa, and others.
Other collagen–vascular disease: Systemic lupus erythematosus, polyarteritis nodosa, and others.
 Hypercoagulable state: Polycythemia, multiple myeloma, cryoglobulinemia, Waldenström macroglobulinemia, antiphospholipid syndrome, factor V Leiden, activated protein C resistance, hyperhomocysteinemia, protein C and S deficiency, antithrombin III mutation, prothrombin mutation G20210A, etc.
Hypercoagulable state: Polycythemia, multiple myeloma, cryoglobulinemia, Waldenström macroglobulinemia, antiphospholipid syndrome, factor V Leiden, activated protein C resistance, hyperhomocysteinemia, protein C and S deficiency, antithrombin III mutation, prothrombin mutation G20210A, etc.
 Rare causes: Migraine, Behçet disease, syphilis, sickle cell disease.
Rare causes: Migraine, Behçet disease, syphilis, sickle cell disease.
 Trauma.
Trauma.
Work-Up
1. Immediate ESR, CRP, and platelets to rule out GCA if the patient is 55 years of age or older. If the patient’s history, laboratories, or both are consistent with GCA, start high-dose systemic steroids. See 10.17, Arteritic Ischemic Optic Neuropathy (Giant Cell Arteritis).
2. Check the blood pressure.
3. Other blood tests: Fasting blood sugar, glycosylated hemoglobin, complete blood count (CBC) with differential, prothrombin time/activated partial thromboplastin time (PT/PTT). In patients younger than 50 years or with appropriate risk factors or positive review of systems, consider lipid profile, antinuclear antibody (ANA), rheumatoid factor, fluorescent treponemal antibody absorbed (FTA-ABS), serum protein electrophoresis, hemoglobin electrophoresis, and further evaluation for hypercoagulable state (see above).
4. Carotid artery evaluation by duplex Doppler US.
5. Cardiac evaluation with electrocardiography (ECG), echocardiography, and possibly Holter monitoring.
6. Consider IVFA, electroretinography (ERG), or both to confirm the diagnosis.
NOTE: It has been suggested that only patients with high-risk characteristics for cardioembolic disease merit echocardiography, including a history of subacute bacterial endocarditis, rheumatic heart disease, mitral valve prolapse, recent myocardial infarction, prosthetic valve, intravenous drug abuse, congenital heart disease, valvular heart disease, any detectable heart murmur, and ECG changes (atrial fibrillation, acute ST segment elevation, or Q waves).
Treatment
There are anecdotal reports of improvement after the following treatments, if instituted within 90 to 120 minutes of the occlusive event. None of these treatments have been proven effective in randomized, controlled clinical trials, and should not be considered the standard of care.
1. Immediate ocular massage with fundus contact lens or digital massage.
2. Anterior chamber paracentesis: See Appendix 13, Anterior Chamber Paracentesis.
3. IOP reduction with acetazolamide, 500 mg i.v., or two 250-mg tablets p.o. or a topical beta-blocker (e.g., timolol or levobunolol, 0.5% b.i.d.).
4. Hyperventilation into a paper bag to induce a respiratory acidosis and subsequent vasodilation.
Follow-Up
1. Refer to an internist for a complete work-up as above.
2. Repeat eye examination in 1 to 4 weeks, checking for neovascularization of the iris/disc/angle/retina (NVI/NVD/NVA/NVE), which develops in up to 20% of patients, at a mean of 4 weeks after onset. If neovascularization develops, perform panretinal photocoagulation (PRP) and/or administer an anti-vascular endothelial growth factor (anti-VEGF) agent.
11.7 BRANCH RETINAL ARTERY OCCLUSION
Symptoms
Unilateral, painless, abrupt loss of partial visual field; a history of transient visual loss (amaurosis fugax) may be elicited.
Signs
(See Figure 11.7.1.)
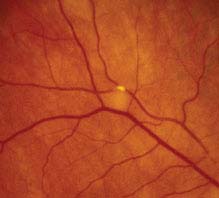
FIGURE 11.7.1. Branch retinal artery occlusion with Hollenhorst plaque.
Critical. Superficial opacification or whitening along the distribution of a branch retinal artery. The affected retina becomes edematous.
Other. Narrowed branch retinal artery; boxcarring, segmentation of the blood column, or emboli are sometimes seen in the affected branch retinal artery. Cholesterol emboli appear as bright, reflective crystals, usually at a vessel bifurcation. Cotton-wool spots may appear in the involved area.
Etiology
See 11.6, Central Retinal Artery Occlusion.
Work-Up
See 11.6, Central Retinal Artery Occlusion. Unlike in CRAO, an ERG is not helpful.
NOTE: When a branch retinal artery occlusion (BRAO) is accompanied by optic nerve edema or retinitis, obtain appropriate serologic testing to rule out cat-scratch disease [Bartonella (Rochalimaea) henselae], syphilis, Lyme disease, and toxoplasmosis.
Treatment
1. No ocular therapy of proven value is available. See treatment in 11.6, Central Retinal Artery Occlusion.
2. Treat any underlying medical problem.
Follow-Up
1. Patients need immediate evaluation to treat any underlying disorders (especially GCA).
2. Reevaluate every 3 to 6 months initially to monitor progression. Ocular neovascularization after branch retinal artery occlusion is rare.
11.8 CENTRAL RETINAL VEIN OCCLUSION
Symptoms
Painless loss of vision, usually unilateral.
Signs
(See Figure 11.8.1.)
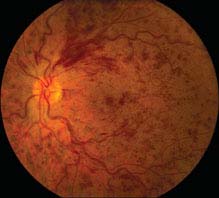
FIGURE 11.8.1. Central retinal vein occlusion.
Critical. Diffuse retinal hemorrhages in all four quadrants of the retina; dilated, tortuous retinal veins.
Other. Cotton-wool spots; disc edema and hemorrhages; macular edema; optociliary collateral vessels on the disc; neovascularization of the optic disc (NVD), retina (NVE), iris (NVI), and angle (NVA).
Differential Diagnosis
 Ocular ischemic syndrome (OIS) or carotid occlusive disease: Dilated and irregular veins without tortuosity. Mid-peripheral retinal hemorrhages are typically present, but disc edema and hemorrhages are not characteristic. Disc neovascularization is present in one-third of cases. Patients may have a history of transient visual loss (amaurosis fugax), transient ischemic attacks, or orbital pain. See 11.11, Ocular Ischemic Syndrome/Carotid Occlusive Disease.
Ocular ischemic syndrome (OIS) or carotid occlusive disease: Dilated and irregular veins without tortuosity. Mid-peripheral retinal hemorrhages are typically present, but disc edema and hemorrhages are not characteristic. Disc neovascularization is present in one-third of cases. Patients may have a history of transient visual loss (amaurosis fugax), transient ischemic attacks, or orbital pain. See 11.11, Ocular Ischemic Syndrome/Carotid Occlusive Disease.
 Diabetic retinopathy: Hemorrhages and microaneurysms concentrated in the posterior pole. Typically bilateral. IVFA differentiates this condition from CRVO. See 11.12, Diabetic Retinopathy.
Diabetic retinopathy: Hemorrhages and microaneurysms concentrated in the posterior pole. Typically bilateral. IVFA differentiates this condition from CRVO. See 11.12, Diabetic Retinopathy.
 Papilledema: Bilateral disc swelling with flame-shaped hemorrhages surrounding the disc. See 10.15, Papilledema.
Papilledema: Bilateral disc swelling with flame-shaped hemorrhages surrounding the disc. See 10.15, Papilledema.
 Radiation retinopathy: History of irradiation. Disc swelling with radiation papillopathy, and retinal neovascularization may be present. Generally, cotton-wool spots are a more prominent feature than hemorrhages.
Radiation retinopathy: History of irradiation. Disc swelling with radiation papillopathy, and retinal neovascularization may be present. Generally, cotton-wool spots are a more prominent feature than hemorrhages.
Etiology
 Atherosclerosis of the adjacent central retinal artery: The artery compresses the central retinal vein in the region of the lamina cribrosa, secondarily inducing thrombosis in the lumen of the vein.
Atherosclerosis of the adjacent central retinal artery: The artery compresses the central retinal vein in the region of the lamina cribrosa, secondarily inducing thrombosis in the lumen of the vein.
 HTN: Most common systemic disease associated with CRVO.
HTN: Most common systemic disease associated with CRVO.
 Optic disc edema.
Optic disc edema.
 Glaucoma: Most common ocular disease associated with CRVO.
Glaucoma: Most common ocular disease associated with CRVO.
 Optic disc drusen.
Optic disc drusen.
 Hypercoagulable state: Polycythemia, multiple myeloma, cryoglobulinemia, Waldenström macroglobulinemia, antiphospholipid syndrome, factor V Leiden, activated protein C resistance, hyperhomocysteinemia, protein C and S deficiency, antithrombin III mutation, prothrombin mutation G20210A, and others.
Hypercoagulable state: Polycythemia, multiple myeloma, cryoglobulinemia, Waldenström macroglobulinemia, antiphospholipid syndrome, factor V Leiden, activated protein C resistance, hyperhomocysteinemia, protein C and S deficiency, antithrombin III mutation, prothrombin mutation G20210A, and others.
 Vasculitis: Sarcoidosis, syphilis, systemic lupus erythematosus, and others.
Vasculitis: Sarcoidosis, syphilis, systemic lupus erythematosus, and others.
 Drugs: Oral contraceptives, diuretics, and others.
Drugs: Oral contraceptives, diuretics, and others.
 Abnormal platelet function.
Abnormal platelet function.
 Orbital disease: Thyroid eye disease, orbital tumors, arteriovenous fistula, and others.
Orbital disease: Thyroid eye disease, orbital tumors, arteriovenous fistula, and others.
 Migraine: Rare.
Migraine: Rare.
Types
 Ischemic CRVO: Multiple cotton-wool spots (usually ≥6), extensive retinal hemorrhage, and widespread capillary nonperfusion on IVFA. RAPD often present and visual acuity is typically 20/400 or worse with visual field constriction. ERG shows decreased b-wave amplitude.
Ischemic CRVO: Multiple cotton-wool spots (usually ≥6), extensive retinal hemorrhage, and widespread capillary nonperfusion on IVFA. RAPD often present and visual acuity is typically 20/400 or worse with visual field constriction. ERG shows decreased b-wave amplitude.
 Nonischemic CRVO: Mild fundus changes. No RAPD is present and acuity is often better than 20/400.
Nonischemic CRVO: Mild fundus changes. No RAPD is present and acuity is often better than 20/400.
Work-Up
Ocular
1. Complete ocular examination, including IOP measurement, careful slit-lamp examination and gonioscopy to rule out NVI and NVA (both of which are best observed before dilation), and dilated fundus examination.
2. IVFA: Risk of neovascularization proportional to degree of capillary nonperfusion.
3. Optical Coherence Tomography: Used to help detect presence and extent of macular edema as well as to monitor response to therapy.
4. If the diagnosis is uncertain, oculopneumoplethysmography or ophthalmodynamometry may help to distinguish CRVO from carotid disease. Ophthalmic artery pressure is low in carotid disease but is normal to increased in CRVO.
Systemic
1. History: Medical problems, medications, eye diseases?
2. Check blood pressure.
3. Blood tests: Fasting blood sugar, glycosylated hemoglobin, CBC with differential, platelets, PT/PTT, ESR, lipid profile.
4. If clinically indicated, particularly in younger patients, consider hemoglobin electrophoresis, VDRL or RPR, FTA-ABS, ANA, cryoglobulins, antiphospholipid antibodies, factor V Leiden mutation, protein C and S levels, antithrombin III mutation, prothrombin G20210A mutation, homocysteine levels, serum protein electrophoresis, and chest radiograph.
5. Complete medical evaluation, with careful attention to cardiovascular disease or hypercoagulability.
Treatment
1. Discontinue oral contraceptives; change diuretics to other antihypertensive medications if possible.
2. Reduce IOP if increased in either eye. See 9.1, Primary Open-Angle Glaucoma.
3. Treat underlying medical disorders.
4. If NVI or NVA is present, perform PRP promptly. Consider PRP if NVD or retinal neovascularization is present. Prophylactic PRP is usually not recommended unless follow-up is in doubt. Intravitreal VEGF inhibitors are very effective in temporarily halting or reversing anterior and posterior segment neovascularization. They may be a useful adjunct to PRP, particularly when rapid reversal of neovascularization is needed.
5. Aspirin 81 to 325 mg p.o. q.d. is often recommended, but no clinical trials have demonstrated efficacy to date and it may increase the risk of hemorrhage.
CRVO-Related Macular Edema
1. Intravitreal ranibizumab 0.5 mg is U.S. Food and Drug Administration (FDA)-approved for treating RVO-related macular edema (ME). Phase III clinical trials have shown promise for the new intravitreal anti-VEGF agent, aflibercept, for the treatment of RVO-related ME. Intravitreal bevacizumab has been used off-label in a similar fashion. Risks of intravitreal injections are low, but include VH and endophthalmitis, among others.
2. Ozurdex, a biodegradable 0.7 mg dexamethasone implant, is FDA-approved for the treatment of ME associated with retinal vein occlusion. Complications include cataract formation and elevated IOP (typically manageable with medical therapy alone).
3. Intravitreal triamcinolone, in both 1 mg and 4 mg doses, is effective in both improving vision and reducing vision loss in patients with ME secondary to CRVO
NOTE: In a large, prospective, randomized trial (SCORE CRVO), a 1 mg dose of intravitreal triamcinolone was found to be equally as effective as a 4 mg dose, but with fewer side-effects (elevated IOP and cataract formation).
Follow-Up
1. Visual acuity 20/40 or better: Every 1 to 2 months for the first 6 months, with a gradual interval taper to annual follow-ups.
2. Visual acuity <20/200: Depends on the type of treatment employed, but in general, every month for the first 6 months, with a gradual taper based on the patient’s condition.
3. Undilated gonioscopy looking for NVA, followed by careful dilated fundus examination looking for NVD or retinal neovascularization, should be performed at each follow-up visit. Evidence of early NVI or NVA should prompt immediate PRP and/or anti-VEGF therapy and monthly follow-up until stabilized or regressed.
4. Patients should be informed that there is an 8% to 10% risk for the development of a BRVO or CRVO in the fellow eye.
Bibliography
Brown DM, Campochiaro PA, Singh RP, et al. Ranibizumab for macular edema following central retinal vein occlusion: six-month primary end point results of a phase III study. Ophthalmology 2010;117(6):1124–1133.
Haller JA, Bandello F, Belfort R Jr, et al. Randomized sham-controlled trial of dexamethasone intravitreal implant in patients with macular edema due to retinal vein occlusion. Ophthalmology 2010;117(6):1134–1146.
Ip MS, Scott IU, VanVeldhuisen PC, et al. A randomized trial comparing the efficacy and safety of intravitreal triamcinolone with observation to treat vision loss associated with macular edema secondary to central retinal vein occlusion: the Standard Care vs Corticosteroid for Retinal Vein Occlusion (SCORE) study report 5. Arch Ophthalmol 2009;127(9):1101–1114.
11.9 BRANCH RETINAL VEIN OCCLUSION
Symptoms
Blind spot in the visual field or loss of vision, usually unilateral.
Signs
(See Figure 11.9.1.)
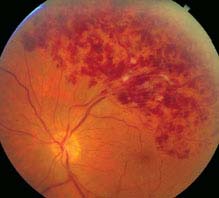
FIGURE 11.9.1. Branch retinal vein occlusion.
Critical. Superficial hemorrhages in a sector of the retina along a retinal vein. The hemorrhages usually do not cross the horizontal raphe (midline).
Other. Cotton-wool spots, retinal edema, a dilated and tortuous retinal vein, narrowing and sheathing of the adjacent artery, retinal neovascularization, VH.
Differential Diagnosis
 Diabetic retinopathy: Dot-blot hemorrhages and microaneurysms extend across the horizontal raphe. Nearly always bilateral. See 11.12, Diabetic Retinopathy.
Diabetic retinopathy: Dot-blot hemorrhages and microaneurysms extend across the horizontal raphe. Nearly always bilateral. See 11.12, Diabetic Retinopathy.
 Hypertensive retinopathy: Narrowed retinal arterioles. Hemorrhages are not confined to a sector of the retina and usually cross the horizontal raphe. Bilateral in most. See 11.10, Hypertensive Retinopathy.
Hypertensive retinopathy: Narrowed retinal arterioles. Hemorrhages are not confined to a sector of the retina and usually cross the horizontal raphe. Bilateral in most. See 11.10, Hypertensive Retinopathy.
Etiology
Disease of the adjacent arterial wall (usually the result of HTN, arteriosclerosis, or diabetes) compresses the venous wall at a crossing point.
Work-Up
1. History: Systemic disease, particularly HTN or diabetes?
2. Complete ocular examination, including dilated retinal examination with indirect ophthalmoscopy to look for retinal neovascularization, and a macular examination with a slit lamp and a 60- or 90-diopter lens, or fundus contact lens to detect ME.
3. Optical Coherence Tomography: Used to help detect presence and extent of macular edema as well as monitor response to therapy.
4. Check blood pressure.
5. Blood tests: Fasting blood sugar, CBC with differential and platelets, PT/PTT, and ESR. If clinically indicated, consider a more comprehensive work-up. See 11.8, Central Retinal Vein Occlusion.
6. Medical examination: Performed by an internist to check for cardiovascular disease.
7. An IVFA is obtained after the hemorrhages clear or sooner if neovascularization is suspected.
Treatment
1. Retinal neovascularization: Sector PRP to the ischemic area, which corresponds to area of capillary nonperfusion on IVFA.
2. Prompt and appropriate treatment of underlying medical conditions (e.g., HTN).
BRVO-Related Macular Edema
1. Focal retinal laser photocoagulation is the traditional gold-standard treatment if edema is present for 3 to 6 months duration, and visual acuity is below 20/40 with macular capillary perfusion. Limitations of focal laser include length of time before effect (often several months) and the need to wait until retinal hemorrhages clear.
2. Intravitreal ranibizumab 0.5 mg is FDA approved for treating RVO-associated ME. Phase III clinical trials have shown promise for the new intravitreal anti-VEGF agent, aflibercept, for the treatment of RVO-related ME. Intravitreal bevacizumab has been used off-label in a similar fashion. Risks of intravitreal injection are low, but include VH and endophthalmitis.
3. Ozurdex implant. See 11.8, Central Retinal Vein Occlusion.
NOTE: There is an evolving trend, particularly in cases of severe edema, to initiate treatment with pharmacologic agents for rapid visual recovery followed by focal laser for better durability of effect.
Follow-Up
In general, every 1 to 2 months for the first 4 months, and then every 3 to 12 months, depending on the treatment employed. At each visit, the patient should be checked for neovascularization and ME.
Bibliography
Campochiaro PA, Heier JS, Feiner L, et al. Ranibizumab for macular edema following branch retinal vein occlusion: six-month primary end point results of a phase III study. Ophthalmology 2010;117(6):1102–1112.
Haller JA, Bandello F, Belfort R Jr, et al. Randomized sham-controlled trial of dexamethasone intravitreal implant in patients with macular edema due to retinal vein occlusion. Ophthalmology 2010;117(6):1134–1146.
11.10 HYPERTENSIVE RETINOPATHY
Symptoms
Usually asymptomatic, although may have decreased vision.
Signs
(See Figure 11.10.1.)
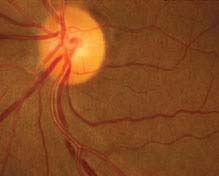
FIGURE 11.10.1. Chronic hypertensive retinopathy with arteriolar narrowing and arteriovenous nicking.
Critical. Generalized or localized retinal arteriolar narrowing, almost always bilateral.
Other
 Chronic HTN: Arteriovenous crossing changes (“AV nicking”), retinal arteriolar sclerosis (“copper” or “silver” wiring), cotton-wool spots, flame-shaped hemorrhages, arterial macroaneurysms, central or branch occlusion of an artery or vein. Rarely, neovascular complications can develop.
Chronic HTN: Arteriovenous crossing changes (“AV nicking”), retinal arteriolar sclerosis (“copper” or “silver” wiring), cotton-wool spots, flame-shaped hemorrhages, arterial macroaneurysms, central or branch occlusion of an artery or vein. Rarely, neovascular complications can develop.
 Acute (“malignant”) HTN: Hard exudates often in a “macular star” configuration, retinal edema, cotton-wool spots, flame-shaped hemorrhages, optic nerve head edema. Rarely serous RD or VH. Areas of focal chorioretinal atrophy [from previous choroidal infarcts (Elschnig spots)] are a sign of past episodes of acute HTN.
Acute (“malignant”) HTN: Hard exudates often in a “macular star” configuration, retinal edema, cotton-wool spots, flame-shaped hemorrhages, optic nerve head edema. Rarely serous RD or VH. Areas of focal chorioretinal atrophy [from previous choroidal infarcts (Elschnig spots)] are a sign of past episodes of acute HTN.
(See Figure 11.10.2.).
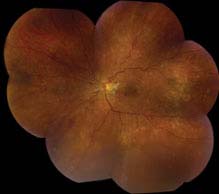
FIGURE 11.10.2. Acute (“malignant”) hypertensive retinopathy.
NOTE: When unilateral, suspect carotid artery obstruction on the side of the normal-appearing eye, sparing the retina from the effects of the HTN.
Differential Diagnosis
 Diabetic retinopathy: Hemorrhages are usually dot-blot, microaneurysms are common, vessel attenuation is less common. See 11.12, Diabetic Retinopathy.
Diabetic retinopathy: Hemorrhages are usually dot-blot, microaneurysms are common, vessel attenuation is less common. See 11.12, Diabetic Retinopathy.
 Collagen–vascular disease: May show multiple cotton-wool spots, but few to no other fundus findings characteristic of HTN.
Collagen–vascular disease: May show multiple cotton-wool spots, but few to no other fundus findings characteristic of HTN.
 Anemia: Mainly hemorrhage without marked arterial changes.
Anemia: Mainly hemorrhage without marked arterial changes.
 Radiation retinopathy: History of irradiation. Most commonly occurs within a few years, but can develop at any time.
Radiation retinopathy: History of irradiation. Most commonly occurs within a few years, but can develop at any time.
 CRVO or BRVO: Unilateral, multiple hemorrhages, venous dilation and tortuosity, no arteriolar narrowing. May be the result of HTN. See 11.8, Central Retinal Vein Occlusion, or 11.9, Branch Retinal Vein Occlusion.
CRVO or BRVO: Unilateral, multiple hemorrhages, venous dilation and tortuosity, no arteriolar narrowing. May be the result of HTN. See 11.8, Central Retinal Vein Occlusion, or 11.9, Branch Retinal Vein Occlusion.
Etiology
 Primary HTN: No known underlying cause.
Primary HTN: No known underlying cause.
 Secondary HTN: Typically the result of preeclampsia/eclampsia, pheochromocytoma, kidney disease, adrenal disease, or coarctation of the aorta.
Secondary HTN: Typically the result of preeclampsia/eclampsia, pheochromocytoma, kidney disease, adrenal disease, or coarctation of the aorta.
Work-Up
1. History: Known HTN, diabetes, or adnexal radiation?
2. Check blood pressure.
3. Complete ocular examination, particularly dilated fundus examination.
4. Refer patient to a medical internist or an emergency department. The urgency depends on the blood pressure reading and whether the patient is symptomatic. As a general rule, a diastolic blood pressure of 110 to 120 mm Hg or the presence of chest pain, difficulty breathing, headache, change in mental status, or blurred vision with optic disc swelling requires immediate medical attention.
Treatment
Control the HTN, as per the internist.
Follow-Up
Every 2 to 3 months at first, and then every 6 to 12 months.
11.11 OCULAR ISCHEMIC SYNDROME/CAROTID OCCLUSIVE DISEASE
Symptoms
Decreased vision, ocular or periorbital pain, after images or prolonged recovery of vision after exposure to bright light, may have a history of transient monocular visual loss (amaurosis fugax). Usually unilateral. Typically occurs in patients who are aged 50 to 80 years. Men outnumber women by 2:1.
Signs
Critical. Although retinal veins are dilated and irregular in caliber, they are typically not tortuous. The retinal arterioles are narrowed. Associated findings include mid-peripheral retinal hemorrhages (80%), iris neovascularization (66%), and posterior segment neovascularization (37%).
Other. Episcleral injection, corneal edema, mild anterior uveitis, neovascular glaucoma, iris atrophy, cataract, retinal microaneurysms, cotton-wool spots, spontaneous pulsations of the central retinal artery, and cherry-red spot. CRAO may occur.
Differential Diagnosis
 CRVO: Similar signs. Decreased vision after exposure to light and orbital pain are not typically found. Ophthalmodynamometry may aid differentiating OIS from CRVO. See 11.8, Central Retinal Vein Occlusion.
CRVO: Similar signs. Decreased vision after exposure to light and orbital pain are not typically found. Ophthalmodynamometry may aid differentiating OIS from CRVO. See 11.8, Central Retinal Vein Occlusion.
 Diabetic retinopathy: Bilateral, usually symmetric. Hard exudates are often present. See 11.12, Diabetic Retinopathy.
Diabetic retinopathy: Bilateral, usually symmetric. Hard exudates are often present. See 11.12, Diabetic Retinopathy.
 Aortic arch disease: Caused by atherosclerosis, syphilis, or Takayasu arteritis. Produces a clinical picture identical to OIS, but usually bilateral. Examination reveals absent arm and neck pulses, cold hands, and spasm of the arm muscles with exercise.
Aortic arch disease: Caused by atherosclerosis, syphilis, or Takayasu arteritis. Produces a clinical picture identical to OIS, but usually bilateral. Examination reveals absent arm and neck pulses, cold hands, and spasm of the arm muscles with exercise.
Etiology
 Carotid disease: Usually ≥90% stenosis.
Carotid disease: Usually ≥90% stenosis.
 Ophthalmic artery disease: Less common.
Ophthalmic artery disease: Less common.
Work-Up
1. History: Previous episodes of transient monocular visual loss? Cold hands or spasm of arm muscles with exercise?
2. Complete ocular examination: Search carefully for neovascularization of the iris, angle, disc, and retina.
3. Medical examination: Evaluate for HTN, diabetes, and atherosclerotic disease. Check pulses. Cardiac and carotid auscultation.
4. Consider IVFA for diagnostic or therapeutic purposes.
5. Noninvasive carotid artery evaluation: Duplex Doppler US, oculoplethysmography, magnetic resonance angiography, others.
6. Consider orbital color Doppler US.
7. Consider ophthalmodynamometry if the diagnosis of CRVO cannot be excluded.
8. Carotid arteriography is reserved for patients in whom surgery is to be performed.
9. Consider a cardiology consultation, given the high association with cardiac disease.
Treatment
Often unsuccessful.
1. Carotid endarterectomy for significant stenosis. Refer to neurovascular surgeon.
2. Consider PRP and anti-VEGF agents in the presence of neovascularization.
3. Manage glaucoma if present. See 9.14, Neovascular Glaucoma.
4. Control HTN, diabetes, and cholesterol. Refer to internist.
5. Lifestyle modification (e.g., smoking cessation).
Follow-Up
Depends on the age, general health of the patient, and the symptoms and signs of disease. Surgical candidates should be evaluated urgently.
11.12 DIABETIC RETINOPATHY
Diabetic Retinopathy Disease Severity Scale
 No apparent retinopathy.
No apparent retinopathy.
 Mild nonproliferative diabetic retinopathy (NPDR): Microaneurysms only.
Mild nonproliferative diabetic retinopathy (NPDR): Microaneurysms only.
 Moderate NPDR: More than mild NPDR, but less than severe NPDR see Figure 11.12.1).
Moderate NPDR: More than mild NPDR, but less than severe NPDR see Figure 11.12.1).
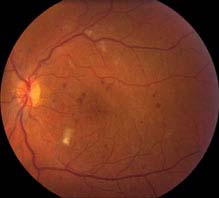
FIGURE 11.12.1. Moderate nonproliferative diabetic retinopathy with microaneurysms and cotton–wool spots.
 Severe NPDR: Any of the following in the absence of proliferative diabetic retinopathy (PDR): diffuse (traditionally >20) intraretinal hemorrhages in all four quadrants, two quadrants of venous beading, or one quadrant of prominent intraretinal microvascular abnormalities (IRMA) (see Figure 11.12.2).
Severe NPDR: Any of the following in the absence of proliferative diabetic retinopathy (PDR): diffuse (traditionally >20) intraretinal hemorrhages in all four quadrants, two quadrants of venous beading, or one quadrant of prominent intraretinal microvascular abnormalities (IRMA) (see Figure 11.12.2).
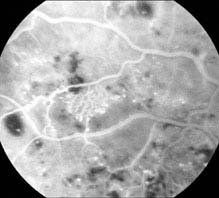
FIGURE 11.12.2. IVFA of intraretinal microvascular abnormality.
 PDR: Neovascularization of one or more of the following: iris, angle, optic disc, or elsewhere; or vitreous/preretinal hemorrhage (see Figures 11.12.3 and 11.12.4).
PDR: Neovascularization of one or more of the following: iris, angle, optic disc, or elsewhere; or vitreous/preretinal hemorrhage (see Figures 11.12.3 and 11.12.4).
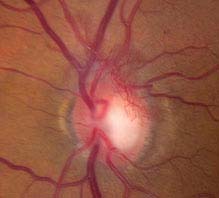
FIGURE 11.12.4. Proliferative diabetic retinopathy with neovascularization of the optic disc.
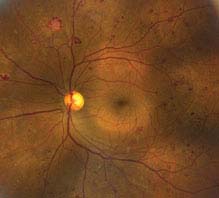
FIGURE 11.12.3. Proliferative diabetic retinopathy with neovascularization and scattered microaneurysms.
 Diabetic macular edema: May be present in any of the stages listed above. Clinically significant macular edema requires treatment and is defined as any one of the following (see Figures 11.12.5 and 11.12.6):
Diabetic macular edema: May be present in any of the stages listed above. Clinically significant macular edema requires treatment and is defined as any one of the following (see Figures 11.12.5 and 11.12.6):
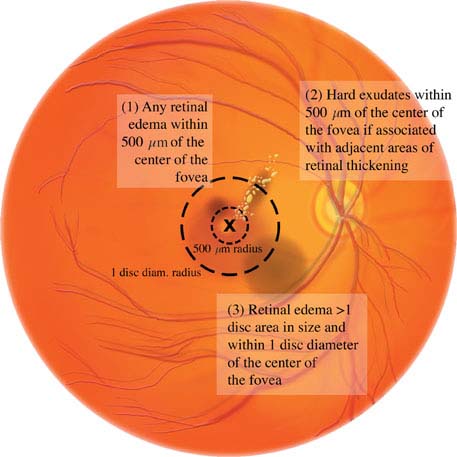
FIGURE 11.12.6. Clinically significant macular edema.
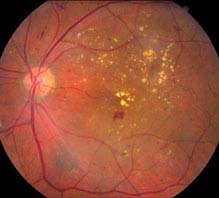
FIGURE 11.12.5. Nonproliferative diabetic retinopathy with clinically significant macular edema.
1. Retinal thickening within 500 mm (one-third of disc diameter) of the foveal center.
2. Hard exudates within 500 mm of the foveal center, if associated with thickening of the adjacent retina.
3. Retinal thickening greater than one disc area in size, part of which is within one disc diameter of the center of the fovea.
Differential Diagnosis for Nonproliferative Diabetic Retinopathy
 CRVO: Optic disc swelling, veins are more dilated and tortuous, hard exudates usually not found, hemorrhages are nearly always in the NFL (“splinter hemorrhages”). CRVO is generally unilateral and of more sudden onset. See 11.8, Central Retinal Vein Occlusion.
CRVO: Optic disc swelling, veins are more dilated and tortuous, hard exudates usually not found, hemorrhages are nearly always in the NFL (“splinter hemorrhages”). CRVO is generally unilateral and of more sudden onset. See 11.8, Central Retinal Vein Occlusion.
 BRVO: Hemorrhages are distributed along a vein and do not cross the horizontal raphe (midline). See 11.9, Branch Retinal Vein Occlusion.
BRVO: Hemorrhages are distributed along a vein and do not cross the horizontal raphe (midline). See 11.9, Branch Retinal Vein Occlusion.
 Ocular ischemic syndrome: Hemorrhages mostly in the mid-periphery and larger; exudates are absent. Usually accompanied by pain; mild anterior chamber reaction; corneal edema; episcleral vascular congestion; a mid-dilated, poorly reactive pupil; iris neovascularization. See 11.11, Ocular Ischemic Syndrome/ Carotid Occlusive Disease.
Ocular ischemic syndrome: Hemorrhages mostly in the mid-periphery and larger; exudates are absent. Usually accompanied by pain; mild anterior chamber reaction; corneal edema; episcleral vascular congestion; a mid-dilated, poorly reactive pupil; iris neovascularization. See 11.11, Ocular Ischemic Syndrome/ Carotid Occlusive Disease.
 Hypertensive retinopathy: Hemorrhages fewer and typically flame-shaped, microaneurysms rare, and arteriolar narrowing present often with arteriovenous crossing changes (“AV nicking”). See 11.10, Hypertensive Retinopathy.
Hypertensive retinopathy: Hemorrhages fewer and typically flame-shaped, microaneurysms rare, and arteriolar narrowing present often with arteriovenous crossing changes (“AV nicking”). See 11.10, Hypertensive Retinopathy.
 Radiation retinopathy: Usually develops within a few years of radiation. Microaneurysms are rarely present. See 11.5, Cotton–Wool Spot.
Radiation retinopathy: Usually develops within a few years of radiation. Microaneurysms are rarely present. See 11.5, Cotton–Wool Spot.
Differential Diagnosis for Proliferative Diabetic Retinopathy
 Neovascular complications of CRAO, CRVO, or BRVO: See specific sections.
Neovascular complications of CRAO, CRVO, or BRVO: See specific sections.
 Sickle cell retinopathy: Peripheral retinal neovascularization. “Sea-fans” of neovascularization present. See 11.20, Sickle Cell Disease (Including Sickle Cell Anemia, Sickle Trait).
Sickle cell retinopathy: Peripheral retinal neovascularization. “Sea-fans” of neovascularization present. See 11.20, Sickle Cell Disease (Including Sickle Cell Anemia, Sickle Trait).
 Embolization from intravenous drug abuse (talc retinopathy): History of intravenous drug abuse, peripheral retinal neovascularization, may see particles of talc in macular vessels. See 11.33, Crystalline Retinopathy.
Embolization from intravenous drug abuse (talc retinopathy): History of intravenous drug abuse, peripheral retinal neovascularization, may see particles of talc in macular vessels. See 11.33, Crystalline Retinopathy.
 Sarcoidosis: May have uveitis, exudates around veins (“candle-wax drippings”), NVE, or systemic findings. See 12.6, Sarcoidosis.
Sarcoidosis: May have uveitis, exudates around veins (“candle-wax drippings”), NVE, or systemic findings. See 12.6, Sarcoidosis.
 Other inflammatory syndromes (e.g., systemic lupus erythematosus).
Other inflammatory syndromes (e.g., systemic lupus erythematosus).
 Ocular ischemic syndrome: See 11.11, Ocular Ischemic Syndrome/Carotid Occlusive Disease.
Ocular ischemic syndrome: See 11.11, Ocular Ischemic Syndrome/Carotid Occlusive Disease.
 Radiation retinopathy: See above.
Radiation retinopathy: See above.
 Hypercoagulable states (e.g., antiphospholipid syndrome).
Hypercoagulable states (e.g., antiphospholipid syndrome).
Work-Up
1. Slit-lamp examination with gonioscopy with careful attention for NVI and NVA, preferably before pharmacologic dilation.
2. Dilated fundus examination by using a 90- or 60-diopter or fundus contact lens with a slit lamp to rule out neovascularization and ME. Use indirect ophthalmoscopy to examine the retinal periphery.
3. Fasting blood sugar, glycosylated hemoglobin, and, if necessary, a glucose tolerance test if the diagnosis is not established.
4. Check the blood pressure.
5. Consider IVFA to determine areas of perfusion abnormalities, foveal ischemia, microaneurysms, and subclinical neovascularization, especially if considering focal macular laser therapy.
6. Consider OCT to evaluate for the presence and extent of ME.
7. Consider blood tests for hyperlipidemia if extensive exudate is present.
Treatment
Clinically Significant Macular Edema
1. Focal or grid laser treatment should be considered in patients with clinically significant macular edema. Patients with enlarged foveal avascular zones on IVFA are treated lightly, away from the regions of foveal ischemia, if they are treated at all. Patients with extensive foveal ischemia are poor laser candidates. Younger patients and diet-controlled diabetic patients tend to have a better response.
2. Patients with diffuse or extensive ME, isolated sub-foveal edema, ME in the presence of foveal ischemia, or those with poor response to prior focal/grid laser may benefit from intravitreal anti-VEGF or corticosteroid therapy, either alone or in combination with laser.
Proliferative Diabetic Retinopathy
Panretinal laser photocoagulation is indicated for any one of the following high-risk characteristics (see Figure 11.12.7):
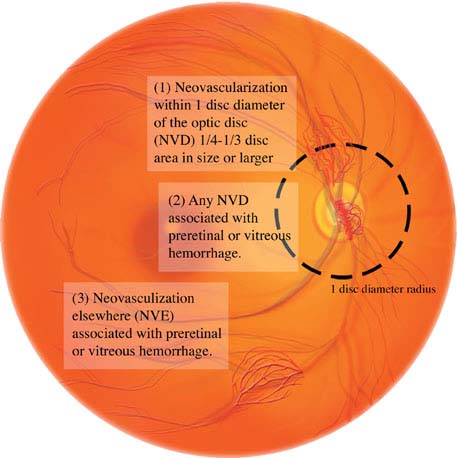
FIGURE 11.12.7. High-risk characteristics for diabetic retinopathy.
1. NVD greater than one-fourth to one-third of the disc area in size.
2. Any degree of NVD when associated with preretinal hemorrhage or VH.
3. NVE greater than one-half of the disc area in size when associated with a preretinal hemorrhage or VH.
4. Any NVI or neovascularization of the angle.
NOTE: Some physicians treat NVE or any degree of NVD without preretinal hemorrhage or VH, especially in unreliable patients. If the ocular media is too hazy for an adequate fundus view, yet one of these criteria is met, anti-VEGF therapy and/or pars plana vitrectomy and endolaser therapy with or without lensectomy and posterior chamber intraocular lens implantation are other options. Anti-VEGF therapy alone must be used cautiously in the presence of retinal traction.
Indications for Vitrectomy
Vitrectomy may be indicated for any one of the following conditions:
1. Dense VH causing decreased vision, especially when present for several months.
2. Traction RD involving and progressing within the macula.
3. Macular epiretinal membranes or recent-onset displacement of the macula.
4. Severe retinal neovascularization and fibrous proliferation that is unresponsive to laser photocoagulation.
5. Dense premacular hemorrhage.
NOTE: Young patients with juvenile type 1 diabetes are known to have more aggressive PDR and therefore may benefit from earlier vitrectomy and laser photocoagulation. B-scan US may be required to rule out tractional detachment of the macula in eyes with dense VH obscuring a fundus view.
Follow-Up
1. Diabetes without retinopathy. Annual dilated examination.
2. Mild NPDR. Dilated examination every 6 to 9 months.
3. Moderate to severe NPDR. Dilated examination every 4 to 6 months.
4. PDR (not meeting high-risk criteria). Dilated examination every 2 to 3 months.
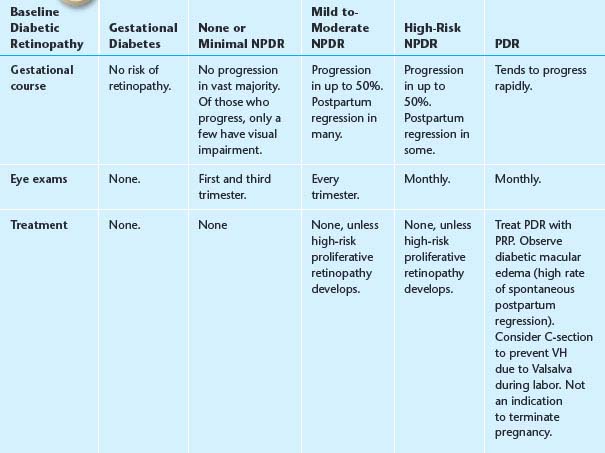
5. Diabetes and pregnancy. Changes that occur during pregnancy have a high likelihood of postpartum regression. See Table 11.12.1 for follow-up recommendations.
TABLE 11.12.1 Recommendations Based on the Baseline Diabetic Retinopathy in Pregnancy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


