Retina & Retinal Disorders
The human retina is the most complex of the ocular tissues with a highly organized structure. It receives the visual image, produced by the optical system of the eye, and converts the light energy into an electrical signal, which undergoes initial processing and is then transmitted through the optic nerve to the visual cortex, where the structural (form, color, and contrast) and spatial (position, depth, and motion) attributes are perceived. The anatomy of the retina is described in Chapter 1, Figure 1–17 showing its layers. Function and functional disturbance in the retina often can be localized to a single layer or a single cell type.
Rod and cone cells in the photoreceptor layer are responsible for the initial transformation, by the process of phototransduction, of light stimuli into the nerve impulses that are conducted through the visual pathways to the visual cortex. These photoreceptors are arranged such that there is an increased density of cones in the center of the macula (fovea), decreasing to the periphery, and a higher density of rods in the periphery. In the foveola, there is a nearly 1:1 relationship between each cone photoreceptor, its ganglion cell, and the emerging nerve fiber, whereas in the peripheral retina, many photoreceptors connect to the same ganglion cell. The fovea is responsible for good spatial resolution (visual acuity) and color vision, both requiring high ambient light (photopic vision) and being best at the foveola, while the remaining retina is utilized primarily for motion, contrast, and night (scotopic) vision.
The rod and cone photoreceptors are located in the avascular outermost layer of the sensory retina. Each rod photoreceptor cell contains rhodopsin, a photosensitive visual pigment embedded in the double-membrane disks of the photoreceptor outer segment. It is made up of two components, an opsin protein combined with a chromophore. The opsin in rhodopsin is scotopsin, which is formed of seven transmembrane helices. It surrounds the chromophore, retinal, which is derived from vitamin A. When rhodopsin absorbs a photon of light, 11-cis retinal is isomerized to all-trans retinal and eventually to all-trans retinol. The resulting configurational change initiates a secondary messenger cascade. Peak light absorption by rhodopsin occurs at approximately 500 nm, which is the blue-green region of the light spectrum. Spectral sensitivity studies of cone photopigments have shown peak wavelength absorption at 430, 540, and 575 nm for blue-, green-, and red-sensitive cones, respectively. The cone photopigments are composed of 11-cis retinal bound to other opsin proteins than scotopsin.
Night (scotopic) vision is mediated entirely by rod photoreceptors. With this dark-adapted form of vision, varying shades of gray are seen, but colors cannot be distinguished. As the retina becomes fully light-adapted, the spectral sensitivity of the retina shifts from a rhodopsin-dominated peak of 500 nm to approximately 560 nm, and color sensation becomes evident. An object takes on color when it selectively reflects or transmits certain wavelengths of light within the visible spectrum (400–700 nm). Daylight (photopic) vision is mediated primarily by cone photoreceptors, and twilight (mesopic) vision by a combination of cones and rods.
The photoreceptors are maintained by the retinal pigment epithelium, which plays an important role in the visual process. It is responsible for phagocytosis of the outer segments of the photoreceptors, transport of vitamins, and reduction of light scatter, as well as providing a selective barrier between the choroid and retina. The basement membrane of the retinal pigment epithelial cells forms the inner layer of Bruch’s membrane, which is otherwise composed of a specialized extracellular matrix and the basement membrane of the choriocapillaris as its outer layer. Retinal pigment epithelial cells have little capacity for regeneration.
Examination of the retina is described in Chapter 2 and depicted in Figures 2–11, 2–12, 2–13, 2–14, 2–15, 2–16 and 2–17. The retina can be examined with a direct or indirect ophthalmoscope or with a slitlamp (biomicroscope) and handheld or contact biomicroscopy lens. This allows identification of the type, level, and extent of retinal disease. Retinal imaging techniques (Figures 2–26, 2–27, 2–28, 2–29 and 2–30) are useful adjuncts to clinical examination, enabling identification of anatomical, vascular (both retinal and choroidal), and functional abnormalities. They include fundus photography, fluorescein angiography, optical coherence tomography (OCT), indocyanine green angiography, and autofluorescence. The clinical application of visual electrophysiologic and psychophysical tests is described in Chapter 2.
Age-related macular degeneration (AMD) affects people aged over 55 and is the leading cause of irreversible blindness in the developed world. It is a complex multifactorial progressive disease. Current evidence suggests genetic susceptibility involving the complement pathway and environmental risk factors, including increasing age, white race, and smoking. Among whites aged over 55, the 10-year risk of developing AMD is approximately 11.5% for early and 1.5% for late disease.
The pathogenesis is still poorly understood; however, degeneration of the retinal pigment epithelium, linked to oxidative stress, seems to be a crucial component. Changes in the adjacent extracellular matrix of Bruch’s membrane and the formation of subretinal deposits are central to disease progression. Progressive diffuse thickening of Bruch’s membrane reduces the ability of oxygen to diffuse through to the retinal pigment epithelium and photoreceptors. The resulting hypoxia results in release of growth factors and cytokines, which stimulate growth of choroidal new vessels. Development of single or multiple areas of weakness in Bruch’s membrane allows the new vessels to grow through into the subretinal space, between the retinal pigment epithelium and the retina, to form a choroidal neovascular membrane. The new vessels leak serous fluid and/or blood, resulting in distortion and reduction of clarity of central vision. Alternatively, visual loss results from progression of the degenerative process to cell death and atrophy of the retinal pigment epithelium.
Twin studies and linkage analysis have identified multiple loci for genes related to AMD. The two most important loci are at 1q25–31 (complement factor H–CFH) and 10q26 (age-related maculopathy susceptibility 2-ARMS2/HTRA serine peptidase 1-HTRA1). The genes can be divided into those which have an influence on structural (HTRA1), inflammatory (CFH, C3, C2, and Factor B), and lipid (APOE) pathways. HTRA1 is a heat shock protein that is involved in the degradation of extracellular proteins such as that found in Bruch’s membrane. Polymorphisms in its promoter gene have been found to be associated with a 10-fold increased risk of AMD. CFH is involved in the alternative complement pathway, thereby identifying an inflammatory component to the pathogenesis of AMD. Its Y402H polymorphism is associated with an increased risk of AMD. C3 mutations confer a 3-fold increased risk, whereas C2 and Factor B appear to have a protective effect. The function of the LOC387715 gene, which is found within the ARMS2 locus next to HTRA1, is unknown but a polymorphism is associated with a 2–3-fold increased risk of AMD, with an additive effect from the CFH polymorphism.
Individuals with genetic predisposition are even more likely to develop the disease if they smoke or have a low intake of antioxidants.
AMD can be classified simply into early and late, the latter being subdivided into geographic atrophy and neovascular disease. The Age-Related Eye Disease Study (AREDS) devised a grading system based on fundal features, of which a simplified form is also useful clinically.
Early AMD is characterized by limited drusen, pigmentary change, or retinal pigment epithelial atrophy. The level of associated visual impairment is variable and may be minimal. Fluorescein angiography demonstrates irregular patterns of retinal pigment epithelial hyperplasia and atrophy.
Drusen are visualized clinically as yellow deposits, which are situated within Bruch’s membrane. They vary in size and shape. They may be discrete or confluent (Figure 10–1). Histopathologically, drusen may also be detected as diffuse subretinal deposits, either basal laminar deposits, formed mainly of collagen-based material and situated between the plasma and basement membranes of the retinal pigment epithelium, or basal linear deposits, consisting of granular lipid-rich material located within Bruch’s membrane.
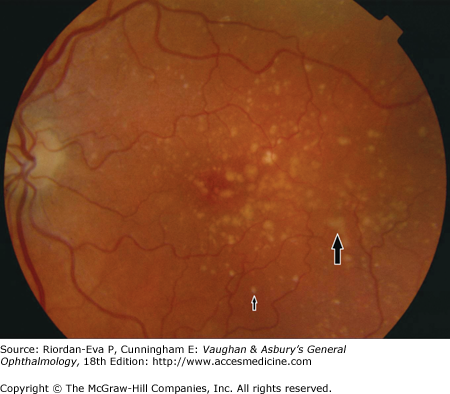
Pigmentary change may be due to focal clumps of pigmented cells in the subretinal space and outer retina, or attenuated areas of hypopigmented retinal pigment epithelium progressing to atrophy.
Geographic atrophy (“dry AMD”) is responsible for up to 20% of legal blindness attributable to AMD. It manifests as well-demarcated areas, larger than two disk diameters, of atrophy of the retinal pigment epithelium and photoreceptor cells, allowing direct visualization of the underlying choroidal vessels. Accumulation of lipofuscin in the retinal pigment epithelium is thought to contribute to the atrophic changes. Visual loss occurs once the fovea is affected. Geographic atrophy is best monitored with autofluorescence imaging, different patterns of abnormality possibly providing clarification of disease progression.
Neovascular (“wet”) AMD is characterized by the development of choroidal neovascularization or serous retinal pigment epithelial detachment. Choroidal new vessels may grow in a flat cartwheel or sea-fan configuration away from their site of entry into the subretinal space to form a choroidal neovascular membrane. Hemorrhagic detachment of the retina may undergo fibrous metaplasia, resulting in an elevated subretinal mass called a disciform scar. Permanent loss of central vision ensues. OCT imaging identifies both subretinal and intraretinal fluid, along with the choroidal neovascular membrane. Fluorescein angiography should be performed on all patients with AMD with new onset of reduced vision or distortion, as the most sensitive method for detection of choroidal neovascularization. It can also guide treatment options. Choroidal neovascularization can be classified angiographically into either classic or occult, depending on the pattern of new vessel growth. Classic is characterized by early hyperfluorescence, which is usually well circumscribed and may have a lacy pattern (Figure 10–2). Occult is characterized by ill-defined and late hyperfluorescence. For research studies, choroidal neovascularization has been subdivided into predominantly classic, in which more than 50% of the lesion has the characteristics of classic choroidal neovascularization; minimally classic, in which less than 50% of the lesion has the characteristics of classic choroidal neovascularization; and pure occult, in which no classic component can be identified.

Retinal pigment epithelial detachment is included in the category of neovascular AMD because of its strong, although not absolute, association with choroidal neovascularization, to the extent that choroidal neovascularization should be assumed to be present until investigations or natural history has excluded it. Serous retinal pigment epithelial detachment may develop from influx of proteinaceous material through a plane of cleavage at the site of drusen. Focal retinal pigment epithelial detachment may also develop from leak of serous fluid from the choroid through small defects in Bruch’s membrane. Retinal pigment epithelial detachments may spontaneously flatten, with variable visual results, but usually leaving an area of geographic atrophy.
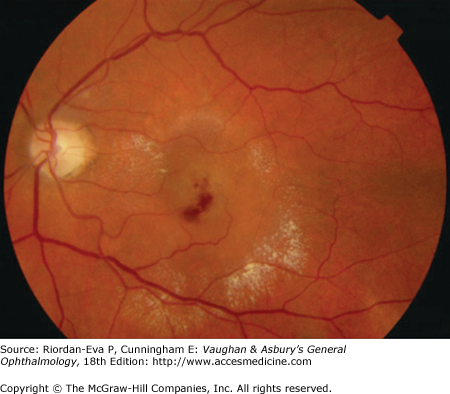
It is uncertain whether retinal angiomatous proliferation (RAP) is a manifestation of AMD, but it usually presents in the same clinical setting. The cause is unknown. It manifests as superficial (inner retinal) hemorrhage with retinal pigment epithelial detachment and extensive exudation (Figure 10–3) and is characterized by anastomosis between the retinal and choroidal circulations (Figure 10–4).

The AREDS, which includes an interventional longitudinal study of progression of AMD, has identified pigmentary changes and large drusen (>250 microns) to be the most important fundal features predictive of progression to late AMD, from which a simple clinical scoring system to predict risk of progression has been devised. Points are assigned according to whether pigmentary changes or large drusen can be identified on fundoscopy. For patients with no late disease, 1 point is assigned for each eye with large drusen, for each eye with pigmentary abnormalities, and if neither eye has large drusen for intermediate-size drusen present in both eyes. For patients with late disease in one eye, 2 points are assigned for the eye with late disease and 1 point for each of large drusen or pigmentary abnormalities in the fellow eye. The 5-year risk of progression to late AMD is 0.5%, 3.0%, 12.0%, 25%, and 50%, respectively, as the cumulative score rises from 0 to 4.
Treatment with oral vitamins and antioxidants, comprising vitamin C (500 g), vitamin E (400 IU), betacarotene (15 mg), and zinc (80 mg) and copper (2 mg) daily, was found in the AREDS to reduce the 5-year risk of progression to late AMD from 28% to 20% in patients with cumulative scores of 3 or 4 on the risk prediction score (see above) but did not show any benefit for those with lower cumulative scores. In a separate study, smokers taking betacarotene have been shown to have an increased risk of development of lung cancer. Therefore, smokers and ex-smokers are advised to omit the betacarotene.
Smoking is a proven risk factor for development of all forms of macular degeneration. Cessation of smoking is thought to reduce the rate of progression, although further trials are required to establish the extent of its effect. It is recommended that smoking be discontinued, together with change in lifestyle to incorporate gentle daily exercise, which lowers the risk of AMD. It takes about 20 years of smoking cessation to reduce the level of risk of development of AMD to that of a non smoker.
Retinal laser photocoagulation reduces the extent of drusen but increases the rate of choroidal neovascularization and is not recommended outside a clinical trial.
Vascular endothelial growth factor (VEGF) plays a crucial role in the expansion of choroidal neovascular membranes. It induces both angiogenesis and increased permeability. Blocking VEGF (anti-VEGF therapy) has become the preferred treatment for neovascular AMD.
Ranibizumab (Lucentis, Genentech) is a humanized Fab fragment of a murine monoclonal anti-VEGF antibody, which is able to bind all isoforms of VEGF. It is able to penetrate through all layers of the retina and is administered by intravitreal injection. The MARINA trial showed stabilization of vision in 94% of eyes with minimally classic or occult lesions and improvement in 34%. The ANCHOR trial showed similar results, with significant benefit over photodynamic therapy (PDT) (see later in the chapter) for predominantly classic lesions.
Currently, ranibizumab is the treatment of choice for all forms of neovascular AMD. Repeated intravitreal injections are well tolerated with minimal side effects, but the ideal treatment regimen is still under investigation. Long-term monthly injections, which are a significant burden to patients and health care systems, may not be needed, but a loading phase of three injections at monthly intervals followed by a maintenance phase of injection frequency being determined by disease activity is probably required. The small PrONTO trial suggested that monthly monitoring with treatment according to results of OCT provides near-equivalent visual outcome to monthly dosing, but this was not confirmed in a larger study using the original retreatment criteria. More rigorous treatment criteria are being investigated to optimize outcome without having to rely upon monthly injections.
The oligonucleotide aptamer (chemically synthesized single-stranded nucleic acid), pegaptanib (Macugen, Eyetech), binds the major pathogenic isoform of VEGF, VEGF165. It is administered by intravitreal injection. Stabilization of vision has been demonstrated in 71% of patients, with improvement occurring in 6% (VISION trial).
Bevacizumab (Avastin, Genentech) is a humanized full-length monoclonal antibody to VEGF. Initially it was thought not to be able to pass through the retina, but it has been widely used with good results. Several (CATT, IVAN, VIBERA and GEFAL) trials are currently being undertaken in the United States and Europe to evaluate its efficacy and safety compared to ranibizumab.
Conventional retinal laser photocoagulation can achieve direct destruction of a choroidal neovascular membrane. It requires confluent high-energy burns over and around the whole membrane. The overlying retina is also destroyed, the laser scar may expand, leading to visual loss, and the rate of recurrence of the neovascular membrane is high. Laser photocoagulation is only used for choroidal neovascular membranes that are more than 200 microns from the center of the foveal avascular zone (extrafoveal).
Photodynamic therapy (PDT) requires an intravenous infusion of a photosensitive dye, verteporfin (Visudyne, Novartis), which is activated by a low-energy visible laser (689 nm). However, this treatment has now largely been replaced by anti-VEGF treatments.
Combining anti-VEGF therapy with PDT steroids, or other agents continues to be investigated. The Mont Blanc study showed that PDT with ranibizumab is no better than ranibizumab alone. Other agents being investigated include the VEGF trap, a designer molecule that binds to VEGF to prevent it binding to its receptor, and RNA interference (RNAi) technology to prevent transcription of VEGF or its receptors.
Surgery for late AMD continues to be studied with mixed results. Options include surgical removal of the choroidal neovascular membrane, macular translocation, and retinal pigment epithelial transplantation. Surgery is recommended only as part of a clinical trial.
Pathologic myopia is one of the leading causes of blindness in the United States and is much more common in the Far East and Japan. It is characterized by progressive elongation of the eye with subsequent thinning and atrophy of the choroid and retinal pigment epithelium in the macula. Usually there is at least 6 diopters myopia. Peripapillary chorioretinal atrophy and linear breaks in Bruch’s membrane (“lacquer cracks”) are characteristic findings on fundoscopy (Figure 10–5). Degenerative changes of the macular pigment epithelium resemble those found in older patients with AMD. A characteristic lesion of pathologic myopia is a raised, circular, pigmented macular lesion called a Fuchs spot. Most patients are in the fifth decade when the degenerative macular changes cause a slowly progressive loss of vision; rapid loss of visual acuity is usually caused by serous and hemorrhagic macular detachment overlying a choroidal neovascular membrane, which occurs in 5%–10% of patients.

Fluorescein angiography shows delayed filling of choroidal and retinal blood vessel and is helpful in identifying and locating the site of choroidal neovascularization in eyes with serous or hemorrhagic detachment of the macula. Anti-VEGF therapy has become the treatment of choice for sub- or juxta-foveal choroidal neovascularization.
The chorioretinal changes of pathologic myopia predispose to retinal breaks and thus to retinal detachment. Peripheral retinal findings may include paving-stone degeneration, pigmentary degeneration, and lattice degeneration. Retinal breaks usually occur in areas involved with chorioretinal lesions, but they also arise in areas of apparently normal retina. Some of these breaks, particularly those of the “horseshoe” and round retinal tear type, will progress to rhegmatogenous retinal detachment.
Diabetic retinopathy is one of the leading causes of blindness in the Western world, particularly among individuals of working age. Chronic hyperglycemia, hypertension, hypercholesterolemia, and smoking are all risk factors for development and progression of retinopathy. Young people with type I (insulin-dependent) diabetes do not develop retinopathy for at least 3–5 years after the onset of the systemic disease. Type II (non-insulin-dependent) diabetics may have retinopathy at the time of diagnosis, and it may be the presenting manifestation.
Early detection and treatment of diabetic retinopathy is essential. Readily detectable changes occur before vision is affected. Their identification and appropriate treatment will usually prevent permanent visual loss. Screening for diabetic retinopathy should be performed within 3 years from diagnosis in type I diabetes, on diagnosis in type II diabetes, and annually thereafter in both types. Digital fundal photography has been proven to be an effective and sensitive method for screening. Seven-field photography is the gold standard, but two 45° fields, one centered on the macula and the other centered on the disk, are becoming the method of choice in most screening programs. Mydriasis is necessary for best quality photographs, especially if there is cataract.
Diabetic retinopathy can progress rapidly during pregnancy. Every pregnant diabetic woman should be examined by an ophthalmologist or digital fundal photography in the first trimester and at least every 3 months until delivery.
Diabetic retinopathy can be classified into nonproliferative retinopathy, maculopathy, and proliferative retinopathy.
Diabetic retinopathy is a progressive microangiopathy characterized by small-vessel damage and occlusion. The earliest pathologic changes are thickening of the capillary endothelial basement membrane and reduction of the number of pericytes. The capillaries develop tiny dot-like outpouchings called microaneurysms. Flame-shaped hemorrhages are so shaped because of their location within the horizontally oriented nerve fiber layer.
Mild nonproliferative retinopathy is characterized by at least one microaneurysm. In moderate nonproliferative retinopathy, there are extensive microaneurysms, intraretinal hemorrhages, venous beading, and/or cotton wool spots (Figure 10–6). Severe nonproliferative retinopathy is characterized by cotton-wool spots, venous beading, and intraretinal microvascular abnormalities (IRMA). It can be diagnosed when there are intraretinal hemorrhages in four quadrants, venous beading in two quadrants, or severe IRMA in one quadrant.

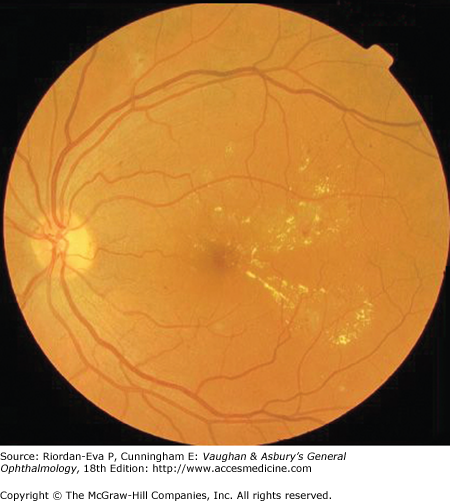
Diabetic maculopathy manifests as focal or diffuse retinal thickening or edema, caused primarily by a breakdown of the inner blood–retinal barrier at the level of the retinal capillary endothelium, which allows leakage of fluid and plasma constituents into the surrounding retina. It is more common in type II diabetes and requires treatment once it becomes clinically significant (Figure 10–7), which is defined as any retinal thickening within 500 microns of the fovea, hard exudates within 500 microns of the fovea associated with retinal thickening, or retinal thickening greater than one disc diameter in size, of which any part lies within one disc diameter of the fovea.
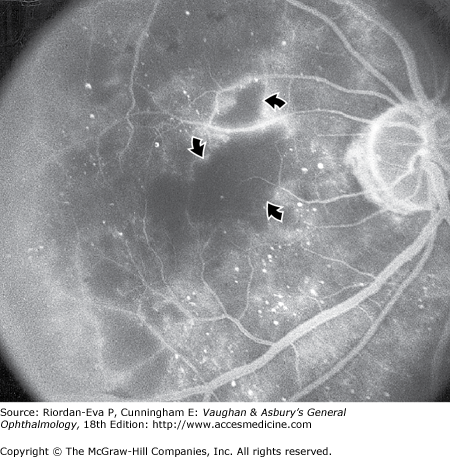
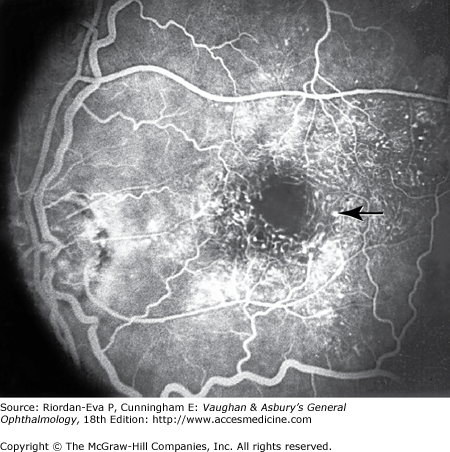
Maculopathy can also be due to ischemia, which is characterized by macular edema, deep hemorrhages, and little exudation. Fluorescein angiography shows loss of retinal capillaries with enlargement of the foveal avascular zone (Figure 10–8).
The most severe ocular complications of diabetes mellitus are due to proliferative diabetic retinopathy. Progressive retinal ischemia eventually stimulates the formation of delicate new vessels that leak serum proteins (and fluorescein) profusely (Figures 10–9 and 10–10). Early proliferative diabetic retinopathy is characterized by the presence of any new vessels on the optic disk (NVD) or elsewhere in the retina (NVE). High-risk characteristics are defined as new vessels on the optic disk extending more than one-third disk diameter, any new vessels on the optic disk with associated vitreous hemorrhage, or new vessels elsewhere in the retina extending more than one-half disk diameter with associated vitreous hemorrhage.


The fragile new vessels proliferate onto the posterior face of the vitreous and become elevated once the vitreous starts to contract away from the retina. If the vessels bleed, massive vitreous hemorrhage may cause sudden visual loss (Figure 10–11). There is a risk of developing neovascularization and vitreous hemorrhage once a complete posterior vitreous detachment has developed. In eyes with proliferative diabetic retinopathy and persistent vitreoretinal adhesions, elevated neovascular fronds may undergo fibrous change and form tight fibrovascular bands, which cause vitreoretinal traction. This can lead to either progressive traction retinal detachment or, if a retinal tear is produced, rhegmatogenous retinal detachment. The retinal detachment may be heralded or concealed by vitreous hemorrhage. When vitreous contraction is complete in these eyes, proliferative retinopathy tends to enter the burned-out or “involutional” stage. Advanced diabetic eye disease may also be complicated by iris neovascularization (rubeosis iridis) and neovascular glaucoma.

Proliferative retinopathy develops in 50% of type I diabetics within 15 years of onset of their systemic disease. It is less prevalent in type II diabetics, but as there are more patients with type II diabetes, more patients with proliferative retinopathy have type II than type I diabetes.
OCT is invaluable in the identification and monitoring of macular edema as well as identification of structural changes within the retina. The development of spectral domain OCT, with increased scan speed and resolution and eye tracking with improved reproducibility, has further enhanced disease assessment and monitoring.
Fluorescein angiography is useful for identifying microvascular abnormalities in diabetic retinopathy (Figure 10–12). Large filling defects of capillary beds—“capillary nonperfusion”—show the extent of retinal ischemia (Figure 10–8) and are usually most prominent in the midperiphery. The fluorescein leakage associated with retinal edema may assume the petaloid configuration of cystoid macular edema (CME) or may be diffuse (Figure 10–13). This can help determine prognosis as well as extent and placement of laser treatment. Eyes with macular edema and significant ischemia have a poorer visual prognosis, with or without laser treatment, than eyes with edema and relatively good perfusion.

The mainstay of prevention of progression of retinopathy is good control of hyperglycemia, systemic hypertension, and hypercholesterolemia.
Ocular treatment depends on the location and severity of the retinopathy. Eyes with diabetic macular edema that is not clinically significant should usually be monitored closely without laser treatment. Clinically significant macular edema requires focal laser if focal and grid laser if diffuse. Argon laser to the macula should be sufficient to produce only light burns, as laser scars can expand and affect vision. Sub-threshold treatment, in which no retinal burn is visualized at the time of treatment, and micropulse laser have been shown to be just as effective with less scarring. Intravitreal injections of triamcinolone or anti-VEGF agents are also effective.
By inducing regression of new vessels, pan-retinal laser photocoagulation (PRP) reduces the incidence of severe visual loss from proliferative diabetic retinopathy by 50%. Several thousand regularly spaced laser burns are applied throughout the retina to reduce the angiogenic stimulus from ischemic areas. The central region bordered by the disk and the major temporal vascular arcades is spared (Chapter 23). Patients at greatest risk of visual loss are those with high risk characteristics. If treatment is delayed until high risk characteristics have developed, it is essential that adequate PRP is achieved without delay. Treatment of severe nonproliferative retinopathy has not been shown to alter the visual outcome; however, if the patient has type II diabetes, poor glycemic control, or cannot be monitored sufficiently carefully, treatment before proliferative disease has developed may be justified.
Vitrectomy is able to clear vitreous hemorrhage and relieve vitreoretinal traction. Once extensive vitreous hemorrhage occurs, 20% of eyes will progress to no perception of light vision within 2 years. Early vitrectomy is indicated for type I diabetics with extensive vitreous hemorrhage and severe, active proliferation and whenever vision in the fellow eye is poor. Otherwise, vitrectomy can be delayed for up to a year as vitreous hemorrhage will clear spontaneously in 20% of eyes. Intravitreal anti-VEGF therapy a few days preoperatively is associated with a reduced re-bleed rate and better visual outcome post operatively. Vitrectomy for proliferative diabetic retinopathy with only mild vitreous hemorrhage is only useful in eyes that have already undergone PRP and have extensive new vessels that have started to fibrose. Tractional retinal detachment does not require vitrectomy until the detachment has involved the fovea. Rhegmatogenous detachment complicating proliferative diabetic retinopathy requires urgent vitrectomy.
Complications following vitrectomy are more common in the type I diabetics undergoing delayed vitrectomy and type II diabetics undergoing early vitrectomy. They include phthisis bulbi, raised intraocular pressure with corneal edema, retinal detachment, and infection.
Retinal vein occlusion is a common and easily diagnosed retinal vascular disorder with potentially blinding complications. The patient presents with sudden painless loss of vision. The clinical appearance varies from a few small scattered retinal hemorrhages and cotton-wool spots to a marked hemorrhagic appearance with both deep and superficial retinal hemorrhage (Figure 10–14), which rarely may break through into the vitreous cavity.

In central retinal vein occlusion the retinal abnormalities involve all four quadrants of the fundus. In branch retinal vein occlusion typically they are confined to one quadrant because the occlusion usually occurs at the site of an arteriovenous crossing (Figure 10–15), but they may involve the upper or lower half (hemispheric branch retinal vein occlusion), or just the macula (macular branch retinal vein occlusion).

Patients are usually over 50 years of age, and more than 50% have associated cardiovascular disease. Predisposing factors and investigations are discussed in Chapter 15. Chronic open-angle glaucoma should always be excluded (see Chapter 11). The major complications associated with retinal vein occlusion are reduced vision from macular edema, neovascular glaucoma secondary to iris neovascularization, and retinal neovascularization.
Macular dysfunction occurs in almost all eyes with central retinal vein occlusion. Although some will show spontaneous improvement, most will have persistent decreased central vision as a result of chronic macular edema, which is also the main cause of persisting impairment of visual acuity in branch retinal vein occlusion.
Macular edema due to central retinal vein occlusion does not respond to laser treatment. In branch retinal vein occlusion, grid-pattern macular argon laser photocoagulation may be indicated when vision loss due to macular edema persists for several months without any spontaneous improvement.
Intravitreal injection of anti-VEGF agents or steroids may be useful. The BRAVO and CRUISE studies, both large multicenter trials, have shown benefit from monthly ranibizumab injections for macular edema secondary to branch and central retinal vein occlusion, respectively. The SCORE study has shown that intravitreal preservative-free triamcinolone provides no benefit compared to laser in branch retinal vein occlusion, but improved outcome compared to observation in central retinal vein occlusion, although it is uncertain whether the optimal dose is 1 mg or 4 mg. Ozurdex (Allergan), an intravitreal implant containing 0.7 mg dexamethasone in a solid polymer drug delivery system, which can be injected into the vitreous using a 22 gauge needle and dissolves completely, has been shown to accelerate improvement in visual acuity compared to placebo in macular edema due to branch or central retinal vein occlusion. The most commonly reported adverse events during the first 6 months after treatment included increased intraocular pressure, but for which only 0.7% of patients required laser or surgical procedures.
Nearly one-third of central retinal vein occlusions are ischemic with significant retinal capillary nonperfusion seen on fluorescein angiography; one-half of these will develop neovascular glaucoma. The standard treatment for iris neovascularization is PRP, although it may also be controlled with intravitreal anti-VEGF agents.
In branch retinal vein occlusion, retinal neovascularization may develop if retinal capillary nonperfusion exceeds five disk diameters in area. Sectoral retinal laser photocoagulation to the area of ischemic retina reduces the risk of vitreous hemorrhage by one-half.
Clinical trials continue to investigate the role of vitrectomy, with or without arteriovenous sheathotomy, to facilitate reperfusion of the retina and reduction of macular edema.
Central retinal artery occlusion causes painless catastrophic visual loss occurring over a period of seconds; antecedent transient visual loss (amaurosis fugax) may be reported. Visual acuity ranges between counting fingers and light perception in 90% of eyes at initial examination. Twenty-five percent of eyes have cilioretinal arteries that continue to perfuse the macula, potentially preserving central vision. An afferent pupillary defect can appear within seconds, preceding any fundus abnormalities, which include opacification of the superficial retina due to infarction, and reduced blood flow in the retinal vessels, possibly manifesting as segmentation of the blood column in the retinal arterioles. A foveal cherry-red spot (Figure 10–16) develops due to preservation of the relatively normal appearance of the choroidal pigment and retinal pigment epithelium through the extremely thin retina overlying the foveola, surrounded by the pale swollen retina of the rest of the macula. The fundal abnormalities resolve within 4–6 weeks, leaving a pale optic disk as the major ocular finding. In older patients, giant cell arteritis must be excluded and if necessary treated immediately with high-dose systemic corticosteroids. Other causes of central retinal artery occlusion are arteriosclerosis and emboli from carotid or cardiac sources. These are discussed further in Chapter 15.

Branch retinal artery occlusion also causes sudden painless visual loss but usually manifesting as impairment of visual field. Visual acuity is only reduced if there is foveal involvement. The extent of the fundal abnormalities, primarily retinal opacification as in central retinal artery occlusion but sometimes accompanied by cotton-wool spots along its border, is determined by the extent of retinal infarction. The cause is often embolic disease, for which clinical evaluation and investigations need to be undertaken (see Chapter 15).
Irreversible retinal damage occurs within a few hours of complete central retinal artery occlusion. Sudden decrease in intraocular pressure resulting in increased retinal perfusion can be achieved with anterior chamber paracentesis and intravenous acetazolamide. This is particularly indicated in embolic central retinal artery occlusion. Inhaled oxygen–carbon dioxide mixture induces retinal vasodilation and increases the Po2 at the retinal surface. Thrombolytic therapy, infused directly into the ophthalmic artery or administered systemically, has been shown to be beneficial but patients rarely present sufficiently early to warrant treatment. Systemic anticoagulants are generally not employed unless necessitated by an embolic cause, such as atrial fibrillation.
Retinal macroaneurysms are fusiform or round dilations of retinal arterioles occurring within the first three orders of arteriolar bifurcation. Most cases are unilateral, involving the superotemporal artery. Two-thirds of patients have associated systemic hypertension.
Macroaneurysms may result in retinal edema, exudation, or hemorrhage typically with an “hourglass” configuration due to bleeding deep and superficial to the retina. Hemorrhage is usually followed by fibrosis of the macroaneurysm such that no treatment is required. If edema threatens the macula, the macroaneurysm can be treated by confluent laser photocoagulation followed by a direct hit. There is a risk that this direct hit will result in hemorrhage, but this usually settles with fibrosis of the macroaneurysm.
Retinopathy of prematurity (ROP) is a vasoproliferative retinopathy that affects premature and low-birth-weight infants. The etiology, classification (Table 10–1), screening regime, and treatment are also discussed in Chapter 17.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


