Purpose
To compare the efficacy and safety of tafluprost, a preservative-free (PF) prostaglandin analogue, with PF timolol in patients with open-angle glaucoma or ocular hypertension.
Design
Randomized, double-masked, multicenter clinical trial.
Methods
After discontinuation and washout of existing ocular hypotensive treatment, patients who had intraocular pressure (IOP) ≥23 and ≤36 mm Hg in at least 1 eye at the 08:00 hour time point were randomized 1:1 to 12 weeks of treatment with either PF tafluprost 0.0015% or PF timolol 0.5%. IOP was measured 3 times during the day (08:00, 10:00, 16:00 hours) at baseline and at weeks 2, 6, and 12. It was hypothesized that PF tafluprost would be noninferior to PF timolol over 12 weeks with regard to change from baseline IOP. The trial was powered for a noninferiority margin of 1.5 mm Hg at each of the 9 time points assessed.
Results
A total of 643 patients were randomized and 618 completed (PF tafluprost = 306, PF timolol = 312). IOPs at the 3 time points assessed during the baseline visit ranged from 23.8 to 26.1 mm Hg in the PF tafluprost group and 23.5 to 26.0 mm Hg in the PF timolol group. IOPs at the 3 time points assessed during the 12-week visit ranged from 17.4 to 18.6 mm Hg for PF tafluprost and 17.9 to 18.5 mm Hg for PF timolol. At all 9 time points, the upper limits of the 2-sided 95% confidence intervals for the difference between treatments in IOP lowering were less than the prespecified noninferiority margin. Similar percentages of PF tafluprost and PF timolol patients reported ocular pain/stinging/irritation (4.4% vs 4.6%) and pruritus (2.5% vs 1.5%). The percentages of PF tafluprost and PF timolol patients reporting conjunctival hyperemia were 4.4% vs 1.2% (nominal P = .016).
Conclusions
The IOP-lowering effect of PF tafluprost was noninferior to that of PF timolol. PF tafluprost is an efficacious and generally well-tolerated ocular hypotensive agent.
Prostaglandin analogues are a relatively new class of ocular hypotensive medications that have proven effective in lowering intraocular pressure (IOP), the major modifiable risk factor implicated in glaucomatous optic nerve damage and chronic visual loss. Prostaglandin analogues act through increased uveoscleral outflow, allowing for IOP reductions that can be additive to the effect of medications that lower IOP through other mechanisms.
The first prostaglandin analogue available in the United States was latanoprost, which has been shown to be effective in reducing IOP but has been associated with a number of local side effects, including ocular hyperemia, increased pigmentation of the iris, and changes to the eyelashes. Similar side effects have also been reported for other more recently approved prostaglandin analogues, including isopropyl unoprostone, bimatoprost, and travoprost.
Many topical eye drop formulations have been formulated with variable concentrations of benzalkonium chloride as a preservative. Benzalkonium chloride may be toxic to a variety of tissues of the ocular surface and may play a role in the tolerability of these formulations over time, especially in those patients with comorbid ocular surface disease such as chronic dry eye. In addition, a subset of patients have delayed-type hypersensitivity (allergy) to benzalkonium chloride. These findings have motivated recent efforts to develop preservative-free (PF) formulations of existing treatments, such as timolol and the dorzolamide/timolol fixed combination. To date, no PF formulations of prostaglandin analogues are available in the United States.
Tafluprost is a new prostaglandin analogue that is being developed in a PF formulation for lowering IOP in patients with glaucoma or ocular hypertension. Tafluprost has been marketed since 2008 in Japan and several European countries, but is not yet approved in the United States. Studies on the preservative-containing (PC) formulation of tafluprost have demonstrated that it is effective and well tolerated for reducing IOP. A small, short-duration (4-week), study found that the PF and PC formulations of tafluprost had equivalent efficacy and tolerability. The purpose of the present study was to compare the safety and efficacy of the PF formulation of tafluprost with PF timolol in a large 12-week phase 3 clinical trial conducted primarily in the United States. Timolol, a beta-blocker, was chosen as the comparator against tafluprost because it is an FDA-accepted standard for glaucoma studies with a well-understood efficacy and safety profile. The results of a meta-analysis suggested that prostaglandin analogues were most effective for lowering IOP but timolol was almost as effective.
Methods
Design
This was a phase 3, multicenter, randomized, double-masked, parallel-group, 12-week, active comparator-controlled clinical trial comparing the efficacy and safety of PF tafluprost (0.0015%) and PF timolol maleate (0.5%) in glaucomatous or ocular hypertensive patients. The trial (Merck Protocol 001) was conducted from February 16, 2010 through September 17, 2010 at 40 sites in the United States, 6 sites in Spain, and 4 sites in Switzerland. A Scientific Advisory Committee composed of non-Merck and Merck scientists contributed to the development of the protocol, statistical analysis plan, analysis and interpretation of the data, and authoring of the manuscript.
Patients
Men and women 18 years and older who had a diagnosis of primary open-angle glaucoma, pigmentary glaucoma, capsular glaucoma/pseudoexfoliation, or ocular hypertension were eligible. Patients on a stable ocular hypotensive medication treatment regimen that began at least 30 days prior to the screening visit, and those who were treatment-naïve, were eligible. At the screening visit, patients had to have a best-corrected visual acuity of +0.6 logMAR (Snellen equivalent of 20/80) or better in each eye and a mean (or median) IOP ≤36 mm Hg in both eyes. Following discontinuation of any existing ocular hypotensive treatment (see Procedure), patients had to have a mean (or median) IOP of ≥23 and ≤36 mm Hg in at least 1 eye at the 08:00 hour time point at the baseline visit and <5 mm Hg difference in mean (or median) IOP between eyes at each of 3 time points (08:00, 10:00, 16:00 hours) at the baseline visit. Patients had to be willing to avoid wearing contact lenses during the study. Key exclusion criteria included abnormal corneal sensation, condition preventing tonometry, ocular opacity or insufficient dilation preventing retinal evaluation, narrow anterior chamber angle, inflammatory ocular surface disease, anterior/posterior uveitis (either eye within 6 months of screening), ocular inflammation/infection, progressive retinal disease, significant ocular signs/symptoms, allergic conjunctivitis, significant visual field defect or progressive visual field loss within the last year, history of certain ocular surgeries, ocular medications (other than antiglaucoma medications or topical lubricants) within 1 week of screening, previous use of tafluprost, significant cardiovascular disease, and history of pulmonary disease including asthma.
Procedure
After screening, eligible patients entered a 5-day to 4-week washout period prior to the baseline visit, dependent upon which prior glaucoma treatment they had used prior to screening (4 weeks for prostaglandin analogues and β-adrenergic antagonists, 2 weeks for α- or β-adrenergic agonists, 1 week for carbonic anhydrase inhibitors and cholinergic agonists, 5 days for miotics). Investigators were allowed to use dorzolamide as rescue treatment (up to 1 week before randomization) if, in the investigator’s opinion, a patient’s IOP was getting too high, or if investigators were concerned that that would occur over the 4-week washout. At the baseline visit, patients had to meet the entry criteria of a mean (or median) IOP of ≥23 and ≤36 mm Hg at 08:00 hours and a <5 mm Hg difference in mean (or median) IOP measurement between eyes at 3 time points (08:00, 10:00, 16:00 hours). Patients who met the study requirements were randomized to 1 of the 2 active treatment groups, PF tafluprost (0.0015%) or PF timolol maleate (0.5%), in a 1:1 ratio and were stratified according to baseline IOP (<25 mm Hg or ≥25 mm Hg at 08:00 hours at the baseline visit) and ocular diagnosis (open-angle glaucoma or ocular hypertension). Patients were assigned to treatment using a computer-generated randomized allocation schedule prepared by a statistician at Merck. Personnel at each study site used an interactive voice response system to determine which masked treatment containers should be given to which patient. The 12-week double-masked treatment period began on the day following the baseline visit. Other study visits occurred at weeks 2, 6, 8, and 12. At these visits, IOP was measured at 08:00, 10:00, and 16:00 hours and other ocular assessments were performed, except at week 8 when the patient returned to the clinical study site for study medication resupply and assessment of dosing compliance only. During the post-study period, patients were to be contacted by telephone to assess for serious adverse events at least 14 days after the last dose of study medication (ie, following discontinuation of study medication or after the last study visit at week 12). Study investigators, site staff, patients, and Merck monitoring staff remained masked to treatment allocation throughout the study.
PF tafluprost (0.0015%) and PF timolol (0.5%) ophthalmic solutions were used. Consistent with the current FDA-approved label for glaucoma, timolol was instilled twice daily. All medications were dispensed from identical unit dose containers. Patients randomized to PF timolol received unit dose pouches of PF timolol marked for morning and evening administration. Patients randomized to PF tafluprost received masked PF vehicle in the morning pouches and active PF tafluprost in the evening pouches. Patients were instructed to administer the morning drops at 08:00 hours (±1 hour) and the evening drops at 20:00 hours (±1 hour). Both eyes were to be treated with study medication regardless of whether 1 or both met the IOP criteria unless, in the judgment of the investigator, the nonqualifying eye should not be treated.
Use of the following medications was prohibited during the study: ocular hypotensive medications other than study medications, topical ocular anti-allergy, anti-inflammatory, or anti-infective medications that could interfere with the study; preservative-containing topical ocular lubricants, periocular topical or intravitreal corticosteroids unless associated with cataract surgery; systemic administration of prednisone or equivalent steroids in sufficient doses and/or for sufficient time such that the investigator had concerns about additional ocular exposure to steroids in terms of safety and/or efficacy; potent CYP2D6 inhibitors (bupropion, fluoxetine, paroxetine, and quinidine).
Efficacy Assessments
IOP was measured using a Goldmann applanation tonometer at the screening clinic visit and at 08:00, 10:00, and 16:00 hours at baseline, and at week 2, 6, and 12 clinic visits. For all ophthalmic procedures, right eye measurements were followed by left eye measurements. At each IOP time point, the examiner took 2 consecutive measurements for each eye. If these 2 consecutive measurements differed by ≤2 mm Hg, then the average IOP for each eye was calculated. However, if the 2 consecutive measurements differed by >2 mm Hg, then a third measurement was performed and the median of the 3 measurements was determined. In practice, the median was used for less than 1% of the data. IOP was measured prior to any dilation.
Safety Assessments
Patients were queried at each study visit for any adverse events occurring while dosing with study drug. For each adverse event, the investigator determined, while masked, the severity, seriousness, and likelihood of being related to study medication. Ocular assessments were performed at each study visit except week 8. Ocular abnormalities identified as clinically relevant were reported as adverse events. Ocular assessments included the ETDRS visual acuity test, external and adnexa ocular examination, automated perimetry (Humphrey or Octopus), slit-lamp examination (including fundus examination and hyperemia assessment), Goldmann applanation tonometry, ocular surface examination using fluorescein staining, gonioscopy, and vital signs. In addition to adverse event reports, conjunctival hyperemia was assessed using the Conjunctival Hyperemia Scale (Ora Inc, Andover, Massachusetts, USA). The examiner graded the degree of conjunctival hyperemia in each eye using a 0-4 scale with increments of 0.5 allowed. A set of color photographs was provided displaying examples of grade 0, 1, 2, 3, and 4 conjunctival hyperemia.
Compliance With Treatment
Patients completed a dosing diary, which was reviewed for compliance at each visit in the treatment period. To confirm compliance with the dosing regimen, patients were asked to return to each study visit with their used and unused study drug containers, which were counted by the investigator. For each patient, a compliant day on therapy was defined as one in which both the morning and evening eye drops were administered as directed. Percent compliance was then calculated as: % compliance = (number of days on therapy/number of days should be on therapy) × 100.
Statistical Analysis
The primary hypothesis was that PF tafluprost would be noninferior to PF timolol with respect to IOP change from baseline over 12 weeks of therapy, based on a noninferiority margin of 1. 5 mm Hg. The primary endpoint used to evaluate efficacy was the mean IOP change from baseline at all 9 time points during the study (08:00, 10:00, and 16:00 hours at weeks 2, 6, and 12). Similarity between tafluprost and timolol was assessed by a noninferiority analysis. The eye with the highest baseline IOP was designated as the “study eye” for the primary efficacy analysis. If both eyes had the same IOP at baseline, then the right eye was designated as the “study eye”; if only 1 eye satisfied the entry criteria, then that eye was designated as the “study eye.” The per-protocol population was used for the analysis of efficacy. This included patients who received at least 1 dose of study treatment, had at least 1 efficacy measurement available for the analysis endpoint, and did not commit any protocol violations that may have substantially affected the results of the primary efficacy endpoints. The final determination on protocol violations was made prior to the final unmasking of the database. A supportive analysis using the full-analysis-set population was performed for the primary efficacy endpoint. The full analysis set consisted of all randomized patients who received at least 1 dose of study treatment and had at least 1 posttreatment efficacy measurement available for the analysis (and baseline measurement for change from baseline analyses). Patients were included in the treatment group to which they were randomized for the analysis of efficacy data. Missing IOP data were imputed by carrying the last observation forward from previous treatment visits (for a specific time point). In practice, the percentages of data imputed at week 12 were 7.1% for PF tafluprost and 3.9% for PF timolol. An analysis of covariance (ANCOVA) model was used including terms for treatment and ocular diagnosis (open-angle glaucoma or ocular hypertension) and baseline IOP as a covariate. A separate ANCOVA model was used for each time point (08:00, 10:00, and 16:00 hours at weeks 2, 6, and 12) to estimate the treatment difference and 95% confidence interval (CI) in terms of the mean change from baseline to the time point. Two supportive statistical methods—an analysis of variance (ANOVA) model including a term for treatment and a constrained longitudinal data analysis (cLDA) model including terms for treatment, week, ocular diagnosis, and the interaction of week by treatment—were also used as sensitivity analyses. Data were analyzed using SAS version 9.1 software (SAS Institute, Cary, North Carolina, USA).
A secondary efficacy endpoint was the proportion of patients with a favorable IOP response, defined as ≥25% reduction in diurnal IOP from baseline at weeks 2, 6, and 12. Diurnal IOP was calculated as the mean of the IOPs in the “study eye” at the 3 time points for each clinic visit (08:00, 10:00, 16:00 hours). The proportions and 95% CIs were provided for each treatment group and for the treatment difference using the Miettinen and Nurminen method. An additional endpoint was the mean (95% CI) change from baseline in diurnal IOP at weeks 2, 6, and 12, which was analyzed using the ANCOVA, ANOVA, and cLDA models and a noninferiority margin of 1.5 mm Hg.
Safety and tolerability were primarily assessed by counts and clinical review of adverse events within 14 days after the last dose of treatment (or after discontinuation). The primary population used for safety analyses was the all-patients-as-treated population, which included all patients who received at least 1 dose of study drug. The proportion of patients who reported 1 or more adverse events, a drug-related adverse event, or a serious adverse event; patients who discontinued because of an adverse event; and adverse events reported by at least 4 patients in any treatment group were calculated. The following groupings of adverse events were prespecified as being of special interest: conjunctival hyperemia, ocular pain/stinging/irritation, and ocular pruritus. P values for the between-treatment differences for these adverse events of special interest were calculated using the Miettinen and Nurminen method.
Sample Size
The primary hypothesis was that PF tafluprost would be noninferior to PF timolol with respect to IOP change from baseline over 12 weeks of therapy. The trial was powered for a noninferiority margin of 1.5 mm Hg, and was designed to enroll 620 patients (310 per treatment group) to yield 576 evaluable patients (288 per treatment group). With this sample size, the probability that the upper bound of the 2-sided 95% confidence interval for the between-treatment difference in mean IOP change from baseline (PF tafluprost minus PF timolol maleate) is ≤1.5 mm Hg was greater than 99% at each of the 9 time points during the study (08:00, 10:00, and 16:00 hours at weeks 2, 6, and 12). The power and sample size were based on the following assumptions at each of the 9 time points during the study, which were based on the results from a previous noninferiority study vs timolol: α = 0.025 (1-sided), noninferiority margin = 1.5 mm Hg, true treatment difference = 0 mm Hg, standard deviation = 3.5 mm Hg.
Results
Patient Accounting and Demographics
The trial profile is shown in Supplemental Figure 1 , (available at AJO.com ). Of 643 randomized patients, 618 patients (96.1%) completed the study and 25 patients (3.9%) discontinued early. Main reasons for discontinuation were patient withdrawal (N = 12) or adverse events (N = 7; discussed below). The reasons for discontinuation were similar among the treatment groups.
A total of 31 patients were excluded from the per-protocol population used for the primary efficacy analysis, 21 in the PF tafluprost group and 10 in the PF timolol group. The main reasons why more patients in the PF tafluprost group were excluded were “patient’s medical history may have had an effect on efficacy data” (4 for PF tafluprost vs 0 for PF timolol; the 4 excluded patients all had a history of asthma in violation of the exclusion criteria—if these patients had been assigned to PF timolol they could have stopped taking study treatment because of an increase in asthma symptoms and/or taken steroids to treat asthma, which might have had an effect on IOP) and “patient received incorrect study medication” (3 for PF tafluprost vs 0 for PF timolol).
Characteristics of the randomized patients by treatment group are summarized in Table 1 . Patient characteristics were generally similar among the treatment groups. Approximately 60% of patients had previously used prostaglandin analogues. The specific ocular treatments used prior to the study are summarized in Table 1 . There were no significant differences ( P > .2) in the proportions of patients taking any of the prostaglandins or timolol/timolol maleate prior to enrollment. The same percentage of patients in each treatment group (19/320 [5.9%] for PF tafluprost and 19/323 [5.9%] for PF timolol) had a history of conjunctival hyperemia.
| Characteristic | Preservative-free Tafluprost (N = 320) | Preservative-free Timolol (N = 323) |
|---|---|---|
| Sex | ||
| Male | 137 (42.8) | 131 (40.6) |
| Female | 183 (57.2) | 192 (59.4) |
| Age | ||
| Mean (SD), years | 63.3 (11.7) | 63.3 (11.6) |
| Race | ||
| White | 236 (73.8) | 244 (75.5) |
| Black/African American | 75 (23.4) | 71 (22.0) |
| Asian | 6 (1.9) | 5 (1.5) |
| Other | 3 (0.9) | 3 (0.9) |
| Geographic location | ||
| United States | 266 (83.1) | 260 (80.5) |
| Other | 54 (16.9) | 63 (19.5) |
| Baseline IOP | ||
| <25 mm Hg | 126 (39.4) | 127 (39.3) |
| ≥25 mm Hg | 194 (60.6) | 196 (60.7) |
| Ocular diagnosis | ||
| Open-angle glaucoma b | 193 (60.3) | 194 (60.1) |
| Ocular hypertension | 127 (39.7) | 129 (39.9) |
| Randomization stratum (baseline IOP/ocular diagnosis) | ||
| <25 mm Hg/open-angle glaucoma b | 78 (24.4) | 78 (24.1) |
| <25 mm Hg/ocular hypertension | 48 (15.0) | 49 (15.2) |
| ≥25 mm Hg/open-angle glaucoma b | 115 (35.9) | 116 (35.9) |
| ≥25 mm Hg/ocular hypertension | 79 (24.7) | 80 (24.8) |
| Prior prostaglandin use | ||
| No use | 129 (40.3) | 145 (44.9) |
| Use | 191 (59.7) | 178 (55.1) |
| Prior ophthalmologic medications c | ||
| Bimatoprost | 40 (12.5) | 32 (9.9) |
| Brinzolamide | 36 (11.3) | 28 (8.7) |
| Carboxymethylcellulose sodium | 25 (7.8) | 16 (5.0) |
| Dorzolamide hydrochloride | 47 (14.7) | 47 (14.6) |
| Latanoprost | 118 (36.9) | 107 (33.1) |
| Polyethylene glycol (+) propylene glycol | 19 (5.9) | 11 (3.4) |
| Timolol | 31 (9.7) | 42 (13.0) |
| Timolol maleate | 25 (7.8) | 28 (8.7) |
| Travoprost | 75 (23.4) | 67 (20.7) |
a Values are number (%) of patients except for age, where mean (SD) is given.
b Includes patients with primary open-angle glaucoma, pseudoexfoliative glaucoma, or pigmentary glaucoma.
Treatment Compliance
The mean percentages of days that patients were fully compliant (ie, took the protocol-specified daily dose) were 97.9% for PF tafluprost and 98.1% for PF timolol. The mean duration of exposure to study treatment was 80.6 days in the PF tafluprost group and 82.1 days in the PF timolol group.
Efficacy
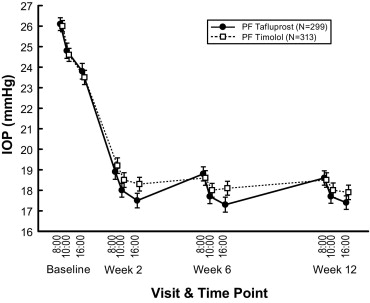
Mean IOPs at each time point during the 12-week study are shown in the Figure . The results for the primary endpoint of mean change from baseline in IOP are shown in Table 2 . The criterion for declaring PF tafluprost noninferior to PF timolol was met since all the upper limits of the 2-sided 95% CIs for the between-group differences in mean change from baseline in IOP were less than the prespecified noninferiority margin of 1.5 mm Hg. Reductions in IOP were observed in both treatment groups as early as week 2. PF tafluprost was noninferior to PF timolol with respect to the IOP change from baseline over the entire 12 weeks of therapy, with the upper limit of the 95% CI for the difference between groups (tafluprost minus timolol) ranging from −0.4 to 0.6 mm Hg at the 9 time points during the study. At 4 of the 9 time points, the upper bounds of the CIs were less than 0, directionally favoring PF tafluprost. Of the 4 instances where the upper limit was less than 0, 3 were at the 16:00 time point on weeks 2, 6, and 12. The between-group differences in mean IOP change from baseline at each time point at week 12 were generally consistent among the subgroups defined by age, race, sex, baseline IOP, and ocular diagnosis ( Supplemental Figure 2 , available at AJO.com ). The results were also similar using an analysis based on the full-analysis-set population, rather than the per-protocol population, and in sensitivity analyses using an ANOVA model and a cLDA model (data not shown).
| Visit | Time Point | Preservative-free Tafluprost | Preservative-free Timolol | Difference a | |||||
|---|---|---|---|---|---|---|---|---|---|
| N | Least Squares Mean (mm Hg) | (95% CI) | N | Least Squares Mean (mm Hg) | (95% CI) | Least Squares Mean (mm Hg) | (95% CI) b | ||
| Week 2 | 08:00 | 280 | −7.1 | (−7.5, −6.8) | 295 | −6.8 | (−7.1, −6.5) | −0.4 | (−0.8, 0.1) c |
| 10:00 | 293 | −6.8 | (−7.1, −6.5) | 305 | −6.1 | (−6.4, −5.8) | −0.7 | (−1.1, −0.3) c | |
| 16:00 | 289 | −6.2 | (−6.5, −5.9) | 302 | −5.3 | (−5.7, −5.0) | −0.8 | (−1.3, −0.4) c | |
| Week 6 | 08:00 | 294 | −7.3 | (−7.6, −6.9) | 308 | −7.4 | (−7.7, −7.1) | 0.1 | (−0.3, 0.6) c |
| 10:00 | 298 | −7.0 | (−7.3, −6.7) | 312 | −6.6 | (−6.9, −6.3) | −0.4 | (−0.9, 0.0) c | |
| 16:00 | 295 | −6.3 | (−6.7, −6.0) | 310 | −5.5 | (−5.9, −5.2) | −0.8 | (−1.3, −0.3) c | |
| Week 12 | 08:00 | 296 | −7.4 | (−7.8, −7.1) | 308 | −7.5 | (−7.8, −7.1) | 0.0 | (−0.4, 0.5) c |
| 10:00 | 298 | −7.0 | (−7.4, −6.7) | 312 | −6.6 | (−7.0, −6.3) | −0.4 | (−0.9, 0.0) c | |
| 16:00 | 295 | −6.2 | (−6.5, −5.9) | 310 | −5.7 | (−6.0, −5.4) | −0.6 | (−1.0, −0.1) c | |
a Difference = Preservative-free tafluprost – preservative-free timolol.
b Based on analysis of covariance with terms for treatment, baseline intraocular pressure, and ocular diagnosis (open-angle glaucoma or ocular hypertension).
c Noninferiority of tafluprost based upon prespecified noninferiority margin of 1.5 mm Hg.
Mean (SD) diurnal IOP at baseline was 24.9 (2.8) mm Hg in the PF tafluprost group and 24.7 (2.5) mm Hg in the PF timolol group. More than half of the patients had ≥25% reduction in diurnal IOP from baseline in both treatment groups (secondary endpoint), and there were numerically more responders in the PF tafluprost group than in the PF timolol group at all 3 visits ( Table 3 ). Mean changes from baseline in diurnal IOP are shown in Table 4 . The criterion for declaring PF tafluprost noninferior to PF timolol with respect to diurnal IOP reduction was met. Directionally, there was a greater mean diurnal IOP reduction from baseline in the PF tafluprost group compared to that of the PF timolol group at all 3 visits, although the upper bound of the 95% CI for the difference was less than 0 only at weeks 2 and 6.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


