Chapter 58 Principles in Surgical Management of Primary Hyperparathyroidism
 This chapter contains additional online-only content, available on expertconsult.com.
This chapter contains additional online-only content, available on expertconsult.com.
In this chapter we discuss the current approach to preoperative workup, review parathyroid surgical anatomy, and detail a comprehensive search algorithm for exploration presented as a general primer and review for the parathyroid surgeon. This information can be seen as standard to parathyroid surgery regardless of the specific surgical approach favored. Readers are also referred to Chapters 59, Standard Bilateral Parathyroid Exploration, 60, Minimally Invasive Single Gland Parathyroid Exploration, 61, Minimally Invasive Video-Assisted Parathyroidectomy, 62, Local Anesthesia for Thyroid and Parathyroid Surgery, 63, Intraoperative PTH Monitoring during Parathyroid Surgery, 64, Radio-Guided Parathyroid Exploration, 65, Surgical Management of Multiglandular Parathyroid Disease, 66, Surgical Management of Secondary and Tertiary Hyperparathyroidism, 67, Parathyroid Management in the MEN Syndromes, and 68, Reoperation for Sporadic Primary Hyperparathyroidism; and for a thorough understanding of parathyroid embryology, see Chapter 2, Applied Embryology of the Thyroid and Parathyroid Glands.
Preoperative Evaluation
Surgical Indications
Primary hyperparathyroidism (primary HPT) can be associated with a wide range of symptoms and complications. These consist of a spectrum of bone disease (bony pain, fractures, osteitis fibrosa cystica), renal (nephrolithiasis, nephrocalcinosis, polyuria, renal insufficiency), gastrointestinal (nausea, vomiting, peptic ulcer disease, constipation, pancreatitis), neuropsychiatric (lethargy, decreased cognitive and social function, depression, psychosis), soft tissue (calcinosis, calciphylaxis, intractable pruritus), and cardiovascular (left ventricular hypertrophy, conduction abnormalities, endothelial dysfunction and decreased QT interval) manifestations.2 Hyperparathyroidism complications have been summarized by the familiar phrase “renal stones, painful bones, abdominal groans, psychic moans (depression), and fatigue overtones.” In most modern series in developed regions of the world, patients generally present with asymptomatic forms of primary HPT or nonspecific symptoms such as fatigue, mild depression, or cognitive impairment, and classic symptoms of hypercalcemia are uncommon.2
Generally, parathyroidectomy benefits symptomatic patients by halting or stabilizing the progression of most complications of primary HPT (see Chapter 56, Primary Hyperparathyroidism: Pathophysiology, Surgical Indications, and Preoperative Workup). Most agree surgery is clearly indicated in symptomatic primary HPT. However, some controversy exists regarding surgical indications in asymptomatic primary HPT. Indications for surgery in asymptomatic primary HPT were originally defined by the first Consensus Development Conference on the Management of Asymptomatic Primary Hyperparathyroidism, sponsored by the National Institutes of Health, in 1990. A second international conference, in 2002, and a third, in May 2008, have revised consensus guidelines for surgical indications in asymptomatic primary HPT.3,4 The most recent (2008) guidelines are summarized and discussed next (see also Table 58-1).

Asymptomatic primary HPT Surgical Guidelines
1. Degree of hypercalcemia. Serum calcium at more than 1 mg/dL above the upper limits of normal.
2. Degree of hypercalciuria. In the absence of renal stones hypercalciuria is no longer regarded as an indication for parathyroid surgery. In the 1990 and 2002 guidelines, hypercalciuria above 400 mg/d was considered an indication for surgery in asymptomatic primary HPT. The basis for this change in recommendation is that hypercalciuria per se has not been established as a risk factor for kidney stones in primary HPT.
3. Renal dysfunction. Glomerular filtration rate (GFR) reduced to < 60 mL/min. In earlier guidelines, surgery was indicated if creatinine clearance was reduced by 30% compared with age-matched normal persons.
4. Bone density reduction. Reduction in the bone density in women and in men over 50, with the T score of –2.5 or less at lumbar spine, femoral neck, total hip, or distal radius. In the 1990 guidelines, forearm Z scores were used.
5. Age. Age less than 50, without symptoms. Surgery is offered to asymptomatic young patients because multiple studies show that approximately 25% of such patients go on to develop one or more of these complications, some of which are irreversible.5 It is not currently possible to determine which young, asymptomatic patients with hyperparathyroidism will go on to develop symptoms.6
6. Other important management factors. Surgery is also indicated in patients for whom medical surveillance and follow-up is difficult, not desired, or not possible.
Controversy has existed regarding surgery for patients older than 50 years of age who are asymptomatic. Talpos, reviewing SF36 Health Survey results, suggested that such so-called asymptomatic patients do, in fact, benefit from surgery.7 Clark has suggested that symptoms of fatigue, joint and bone pain, and depression are frequently present in such supposedly asymptomatic patients and that these symptoms improve with surgery.8 Clark believes that less than 5% of patients with primary hyperparathyroidism are truly asymptomatic.8 Silverberg has shown that not only asymptomatic patients treated with surgery have improved and sustained lumbar spine and femoral neck (cancellous) bone density, but bone density that may have remained stable for 10 years in the presence of hyperparathyroidism (HPT) tends to deteriorate thereafter.9 Successful parathyroidectomy has also been shown to improve muscle strength and fine motor function.10 Zanocco et al. demonstrated that cost-effectiveness analysis showed parathyroid surgery to be the optimal strategy for asymptomatic patients > 50 years of age.11 The trend not only among surgeons, but endocrinologists as well, is a much more “pro-surgery” attitude.
Preoperative History and Physical Examination
History
• Symptoms of hypercalcemia. Weight loss, polydipsia, fatigue, memory change, depression, renal stones, hypertension, bone-joint-muscle pain, arthritis, gout, fracture, bone disease, nausea, vomiting, peptic ulcer, pancreatitis.
• Time course of hypercalcemia:
• Age of onset. Sporadic HPT usually occurs in older age group. Some familial HPT syndromes may present at an earlier age (see the primary HPT genetic section presented later in the chapter).
• History related to drugs and conditions that mimic HPT:
• History of radiation exposure. Low-dose radiation exposure can increase the risk of hyperparathyroidism threefold.12
• Family history. Hypertension, endocrine tumor, calcium disorder.
• Past history. Neck, thyroid, parathyroid surgery.
• History related to specific genetic syndromes. Described fully in genetics section presented later in the chapter.
 Please see the Expert Consult website for more discussion of this topic.
Please see the Expert Consult website for more discussion of this topic.
• Six -question Panel for MEN 1 Assessment. Yip et al. suggested a six-question panel assessment for possible multiple endocrine neoplasia 1 (MEN 1) can be used to screen for MEN 1 in apparent sporadic primary HPT. The assessment question panel includes the following: Have any blood relatives had (1) neck surgery? (2) Kidney stones? (3) Brain tumors? (4) Ulcers? (5) High calcium levels? (6) Pancreatic tumors?
Biochemical screening to rule out MEN 1 was recommended in HPT patients with multiple positive panel answers. It was noted that young males with multiple positive panel answers have higher chances of MEN 1 than females with positive panel scores or males without positive answers. Age and positive panel answers were associated with increased chance of having MEN 1, although male sex alone was not. A false positive result of 23% was noted with question panel positivity.13
Physical Exam
• Physical examination—typically unremarkable. Albright’s dictum states that if a palpable nodule is present in a patient with hyperparathyroidism, it is typically an unrelated thyroid nodule; 50% of parathyroid carcinoma patients may have a palpable mass.14 A firm palpable neck mass in a setting of severe preoperative hypercalcemia and parathyroid hormone (PTH) elevation would strongly suggest the possibility of parathyroid carcinoma.
• Preoperative laryngeal examination—to assess preoperative vocal cord function—is important, especially in the reoperative setting.15,16
Preoperative Lab Work
• Serum calcium and intact parathyroid hormone (IPTH). High calcium and high IPTH basically make the diagnosis of HPT.
• Serum albumin, phosphate, magnesium, and chloride should be measured.
• Alkaline phosphatase. It is elevated in the setting of bone disease. When elevated preoperatively a surgeon should become alert that severe postoperative hypocalcemia, termed hungry bone syndrome, may occur.
• Vitamin D-25-OH vitamin D should be evaluated in patients being assessed for HPT. This has important implications in the differential diagnosis as well as in management (see the section that follows on vitamin D deficiency and HPT). Conditions that can be associated with vitamin D deficiency should be discussed, including Crohn’s disease, cystic fibrosis, celiac disease, malabsorption resulting from gastric bypass surgery, or renal dysfunction (with decreased ability to synthesize the active dihydroxylated form).17
• Assessment of renal function.
• Twenty-four-hour urine calcium and creatinine to assess renal function. This also helps to rule out BFHH (benign familial hypercalciuric hypercalcemia), which can mimic HPT; diagnosis is made with low urine calcium/creatinine clearance ratio and total calcium/24-hour urine collection < 100 mg. Surgeons are well advised to be knowledgeable and astute about the biochemical diagnosis of HPT. In the operating room, surgeons should be fully confident that the diagnosis is accurate, which implies their commitment to explore thoroughly, recognizing that 1 or more abnormal glands will be present and amenable to curative resection through a neck incision about 98% of the time (Box 58-1).
Preoperative Genetic Assessment
Familial Syndromes and Genetic Testing
Primary hyperparathyroidism (HPT) is predominantly sporadic but may be seen as part of an inherited syndrome. For a surgeon, it is important to distinguish preoperatively if a patient with primary hyperparathyroidism is sporadic or syndromic in nature. The diagnosis of syndromic HPT has implications as to the number of glands involved and hence on the aggressiveness of surgery necessary as well as important implications for family screening. Genetic testing is not recommended on a routine basis. Varying penetrance and incomplete syndromic manifestations can make the diagnosis of syndromic HPT challenging. A clinician must look for clues during initial preoperative evaluation to identify when and which genetic tests should be ordered (Table 58-2).
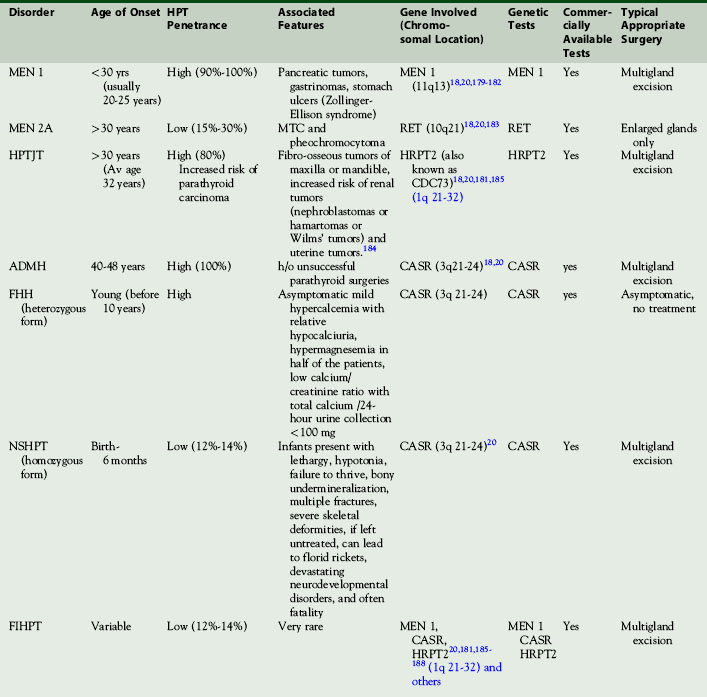
Multiple Endocrine Neoplasia Type 1 (MEN 1)
MEN 1 is an autosomal dominant disorder of the MEN 1 gene (encoding menin) and is also known as Wermer’s syndrome. It presents with tumors predominantly of the parathyroid, pancreatic, and pituitary glands. Hyperparathyroidism is the most frequent endocrine expression of MEN 1, occurring in 95% by age 50. MEN 1–associated HPT presents at earlier age, on average at 20 years, 30 years younger than for typical nonsyndromic primary HPT (see Chapter 67, Parathyroid Management in the MEN Syndromes).18 Yip et al. found that MEN 1 is more common in young males presenting with primary HPT and proposed a six-question panel (described on Expert Consult website).13
Multiple Endocrine Neoplasia Type 2A (MEN 2A)
MEN 2A is an autosomal dominant disorder resulting from mutations in the RET gene. It is also known as Sipple’s syndrome and typically consists of medullary thyroid cancer, pheochromocytoma (in 50%), and HPT (in 10% to 30%) cases (see Chapter 67, Parathyroid Management in the MEN Syndromes).
Autosomal Dominant Mild Hyperparathyroidism (ADMH)
This is a rare autosomal dominant syndrome presenting with hypercalcemia and hypercalciuria. It is associated with CASR gene mutation.19 This syndrome presents in an older age group, usually after 40 years of age, and hyperparathyroidism is seen in all cases. History of failed parathyroid surgery is noted in many patients. Multigland excision is the treatment of choice.20
Familial Hypocalciuric Hypercalcemia (FHH)
This is a rare autosomal dominant disorder presenting with asymptomatic nonprogressive lifelong hypercalcemia with 100% penetrance.21,22 It is typically diagnosed in families by the presence of hypercalcemia and relative hypocalciuria with variable PTH elevation. This represents a renal calcium set point disorder and is not managed with parathyroid surgery but needs to be preoperatively distinguished from primary HPT and is the main reason for checking a 24-hour urine collection for total calcium and creatinine in patients preoperatively. Almost half of FHH patients have increased serum magnesium and a diagnostic low calcium/creatinine clearance ratio (usually below 0.01) with total calcium for a 24-hour urine collection of typically < 100 mg. FHH is the phenotype when this gene is heterozygous, whereas neonatal severe hyperparathyroidism is present phenotypically when the gene is homozygous.
Familial Isolated Hyperparathyroidism (FIHPT)
This is rare autosomal dominant syndrome, which is actually a phenotypic subgroup resulting from incomplete expression of MEN 1, FHH, HPTJT, or some other syndromic cause where syndromic manifestation is limited to HPT. Hannan et al. have reported on two cases of FIHPT, one with symptomatic hypercalcemia and the other asymptomatic.23 They suggest that if a detailed family history shows that first-degree relatives are affected, then genetic analysis should be performed to rule out MEN 1, MEN 2, FIHPT, or BFHH.
History and Physical Examination Findings That May Indicate Genetic Disorder
Although age at onset is not an absolute indicator of syndromic HPT, generally the occurrence of primary hyperparathyroidism in children or young adults suggests the possibility of a familial syndrome. This is especially true of young symptomatic patients such as those with recurrent nephrolithiasis.24
A personal history of jaw tumors, uterine polyps, or parathyroid cancer points to HPTJT syndrome, whereas a history of pituitary adenoma, Zollinger-Ellison syndrome, pancreatic tumors, gastrinoma, stomach ulcers, or the presence of multiple facial angiofibromas strongly suggests MEN 1.25 MEN 2A Sipple’s syndrome is suggested by symptoms or a history of medullary thyroid cancer or pheochromocytoma.
Vitamin D and primary HPT
Second, vitamin D deficiency is common and so may coexist in patients with primary HPT. Interestingly, vitamin D deficiency and insufficiency seem to be more prevalent in patients with primary hyperparathyroidism than in geographically matched populations.26 Primary hyperparathyroidism seems to be more severe in those with concomitant vitamin D deficiency presenting with advanced disease phenotype, at least biochemically.26–30 Coexisting vitamin D deficiency may cause the serum calcium level to fall into the normal range, which may lead to diagnostic uncertainty. The cause of vitamin D deficiency is usually multifactorial, resulting from either inadequate exposure to ultraviolet radiation, inadequate vitamin D intake (dietary), vitamin D malabsorption due to certain medical conditions like Crohn’s disease, cystic fibrosis, celiac disease, or past gastric bypass surgeries, or inability to synthesize the active dihydroxylated compound as in renal dysfunction.17
Vitamin D–deficient patients undergoing parathyroidectomy are also at increased risk of postoperative hypocalcemia and “hungry bone syndrome,” which underscores the importance of a preoperative assessment of vitamin D status in all patients with primary hyperparathyroidism.26 A postoperative rise in PTH is more likely in patients with preoperative vitamin D deficiency. Vitamin D supplementation following parathyroidectomy for primary hyperparathyroidism reduces the incidence of this postoperative eucalcemic PTH elevation.31
It may be difficult to treat vitamin D deficiency in the setting of primary hyperparathyroidism, and close monitoring of serum and urine calcium levels is essential, as limited data suggest that those levels may increase in some patients in response to vitamin D replacement therapy.27 Current guidelines recommend measurement of serum levels of 25-hydroxy vitamin D (25-OH vitamin D) in all patients with primary HPT, and repletion if the levels are less than 50 mmol/L (20 ng/mL). If vitamin D is to be given, it should be done with care, close follow-up, and at low doses. Preliminary data on vitamin D repletion in patients with mild primary hyperparathyroidism suggest that, in some cases, vitamin D deficiency may be corrected without worsening the underlying hypercalcemia.26,30
Normocalcemic Primary Hyperparathyroidism
Normocalcemic primary HPT is a newly recognized entity in which patients have normal total serum calcium concentrations but present with parathyroid hormone levels that are consistently elevated in the absence of a secondary cause of hyperparathyroidism.32 Although total calcium levels are normal, ionized levels may be elevated in these patients.
Patients with true normocalcemic primary HPT are largely asymptomatic. Patients seen in a referral center with normocalcemic hyperparathyroidism have more substantial skeletal involvement than is typical in primary HPT, and over time these changes tend to progress. These patients usually seek medical attention in the context of an evaluation for decreased bone mass.33 A certain percentage of patients who have normocalcemic primary hyperparathyroidism will progress to overt hypercalcemic primary hyperparathyroidism. It seems evident, however, that progression to hypercalcemia is not inevitable, nor is there any uniform time course for the development of hypercalcemia. Many of patients with normocalcemic primary HPT continue to show normal concentrations of the serum calcium over time.32 Some have suggested these patients may represent an early form of symptomatic primary HPT.34 It is suggested that patients with normocalcemic primary HPT should be monitored regularly for progression of their disease. If the disease is worsening, then a proactive, surgical approach would seem to be appropriate.32 Data on normocalcemic primary HPT are limited, and further research on this entity is warranted (see also Chapter 56, Primary Hyperparathyroidism: Pathophysiology, Surgical Indications, and Preoperative Workup).
Localization Testing
 Please see the Expert Consult website for more discussion of this topic. See also Chapter 57, Guide to Preoperative Parathyroiod Localization Testing.
Please see the Expert Consult website for more discussion of this topic. See also Chapter 57, Guide to Preoperative Parathyroiod Localization Testing.
Localization Testing
Localization testing is necessary if one is to offer unilateral or minimal access surgery (see Chapter 57, Guide to Preoperative Parathyroid Localization Testing). Our preference is for a combination of sestamibi and ultrasound analysis. These two studies provide a complementary functional (sestamibi) and structural (ultrasound) assessment and have been shown to be extremely reliable, even in the reoperative setting.35 Sestamibi scanning appears to be the most sensitive preoperative test modality available, and when oblique views plus single-photon emission computed tomography (SPECT) imaging are incorporated, it is often sufficient without the addition of ultrasound. Most researchers feel that it is less accurate in the setting of multiglandular disease, revision surgery, mediastinal lesions, and morbid obesity. Although ultrasound is inexpensive and noninvasive, it is operator dependent and can miss lesions because of acoustic shadowing behind the larynx and trachea and in the mediastinum. Primary hyperparathyroid patients with preoperative positive MIBI and negative ultrasound (US) are more likely to have posterior located upper gland adenomas.36
A four-dimensional computed tomography (4D-CT) may provide greater sensitivity than sestamibi imaging and ultrasonography for precise (quadrant) localization of hyperfunctioning parathyroid glands and should be considered if initial localization studies are negative. For patients undergoing reoperative exploration for recurrent or persistent parathyroid disease, 4D-CT appears to offer improved preoperative planning.37,38
Single photon emission computed tomography/computed tomography (SPECT/CT) offers the advantage of combining function and anatomy for exact localization of ectopic parathyroid adenomas. SPECT/CT may provide more reliable localization of ectopic adenomas.39
Uniglandular versus Multiglandular Disease
In four-gland hyperplasia, all four glands are hyperplastic and some, or all, may be enlarged. The individual glands affected by hyperplasia, as just stated, may vary from normal size to markedly enlarged. Glands affected with hyperplasia, like single-gland adenomas, are hypercellular with decreased fat. Although histologic differences between adenomas and hyperplastic glands have been proposed (adenomas having a normal rim of parathyroid tissue around the adenoma, and hyperplastic glands having a thick capsule, greater cellular atypia, a more mixed cellularity of chief cells, and oncocytes, with gland lobulation and thick-walled vessels), it is generally accepted that adenomas and hyperplastic glands cannot be segregated based on histologic criteria.40 DeLellis has shown that only 50% of known adenomas have the “normal rim” sign.41 Further, some hyperplastic glands affected with nodular hyperplasia may have pseudo rims.42 Given these histologic findings, the diagnosis of single-gland disease versus four-gland hyperplasia has traditionally been made with a combination of gross surgical and histologic findings (see Chapter 70, Surgical Pathology of the Parathyroid Glands).
Double Adenoma
In primary hyperparathyroidism there appears to be a third, distinct, pathologic entity—double adenoma—that takes the middle ground between single-gland adenoma and four-gland hyperplasia. When explored, some patients are found to have only two enlarged glands and are cured upon resection of these two glands alone. Double adenoma may be considered to be a form of asynchronous four-gland hyperplasia, but the high long-term cure rate associated with two-gland resection would argue toward the existence of double adenoma as a separate legitimate entity. Szabo and others have shown that the recurrence rate after initial successful surgery for double adenoma is equivalent to that after successful single-adenoma surgery (approximately 1% to 2%), with both single-gland and double adenoma recurrence rates being significantly lower than for multiglandular disease (approximately 9.2% recurrence rate).43,44 In a series reviewing 1962 patients with primary hyperparathyroidism, Edis reported finding two large glands and two normal-sized glands in 1.9% of patients. These patients were rendered eucalcemic through resection of only the two large glands.45 Most believe that double adenoma accounts for from 2% to 5% of patients with primary hyperparathyroidism.46,47 Some reports describe double adenoma rates as high as 12%.43 Thompson noted the increased frequency of double adenoma in patients over 65 years of age, with a rate of 9% in this age group.48
The incidence of parathyroid carcinoma in patients who have primary hyperparathyroidism is less than 1% (see Chapter 70, Surgical Pathology of the Parathyroid Glands). The term parathyromatosis refers to the proliferation of chief cell rests to form supernumerary and rudimentary glands in some forms of hyperparathyroidism, such as MEN 1, or secondary hyperparathyroidism. Multiple parathyroid gland implants may develop (postsurgical parathyromatosis) with rupture and spillage of parathyroid tumor and may lead to recurrent hyperparathyroidism.49
Asynchronous Multiglandular Disease
The occurrence of asynchronous multiglandular disease is supported by multiple studies that show that the rates of recurrent (not persistent) hyperparathyroidism after initial biochemical cure range from 1% to 16%.50–54 Worsey’s series of 371 patients with 15-year follow-up showed that when disease recurred, it did so on average 3.8 years after an initially successful operation.53 In such patients with asynchronous multiglandular disease, it appears that although they have a four-gland process, the polyclonal hyperplastic tendency is weak and not clinically manifest at least initially and in some may never develop clinically. With this construct in mind, it is interesting to note that several milder forms of primary hyperparathyroidism (including single adenoma and some forms of multiglandular disease, such as double adenoma, hyperparathyroidism in MEN 2A, and sporadic noninherited hyperplasia) are well treated, with low recurrence rates, when only grossly enlarged glands are excised. Thus, in some subset of these patients with a four-gland process, asynchronous changes may be so gradual as to never become clinically manifest. Within other forms of multiglandular disease in primary hyperparathyroidism (such as MEN 1), the tendency for all four glands to proliferate and form clones is considerably stronger. This translates into a much more aggressive clinical course, as evidenced by a 50% recurrence rate in patients with MEN 1 at 12 years after initially successful subtotal parathyroidectomy.55 A higher recurrence rate after aggressive parathyroid exploration and resection is also noted in neonatal hyperparathyroidism, familial hyperparathyroidism, and secondary hyperparathyroidism. Four-gland disease can be thought of as a spectrum of disease with varying degrees of penetrance and clinical aggressiveness (Figure 58-1). Changes may evolve over time, leading to recurrence through asynchronous changes in different glands. In more clinically virulent forms of multiglandular disease, even supernumerary glands develop and become clinically manifest.41,56 Excision of these supernumerary glands may require bilateral thymectomy and resection of adipose tissue surrounding the parathyroid glands.
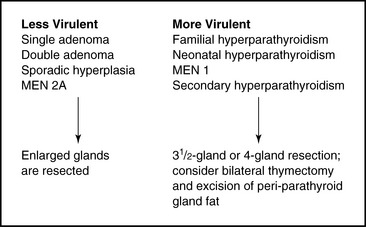
Treating the less virulent forms of multiglandular disease with conservative resection of only grossly enlarged glands results in a high cure rate and is supported by the work of Badder and Harrison, which has emphasized the clinical insignificance of microscopic hyperplasia.57–59 A more aggressive subtotal resection in these patients can lead to rates of hypoparathyroidism of more than 20%.60
Intraoperative PTH: A Functional Criteria for Uniglandular versus Multiglandular Disease
In the past and to some degree today, uniglandular disease was distinguished from multiglandular disease (MGD) with a combination of gross surgical and histologic findings. If the surgeon judged that a given gland was greater than normal size and if the pathologist judged there was increased cellularity and decreased stromal fat, the gland was judged to be diseased and therefore clinically significant. With these gross surgical/histologic criteria, multiglandular disease was judged to be present in approximately 15% of patients with primary hyperparathyroidism. In fact, in the past it was suggested that most patients with primary hyperparathyroidism had hyperplasia and that all these patients should be treated with subtotal parathyroidectomy.61 A new alternate test, intraoperative PTH (iPTH), is available to the surgeon to facilitate the diagnosis of MGD. There are two options for diagnosing MGD after the first enlarged gland is removed: the identification of additional grossly enlarged glands that are histologically hyperplastic, or through intraoperative PTH. The former is the traditional gross and histologic definition approach that requires bilateral exploration and has its advocates and claims rates of MGD of 15% to 30% (see Chapter 59, Standard Bilateral Parathyroid Exploration). The later approach using iPTH as a biochemical test for additional hyperfunctional glands also has its advocates and claims rates of MGD closer to between 4% and 5%62 (see Chapter 63, Intraoperative PTH Monitoring during Parathyroid Surgery). Advocates of both approaches claim success for their respective algorithms. It does appear that with obligatory bilateral exploration some enlarged glands are found but some of these glands may not be functionally active and their excision may not be necessary because iPTH-guided parathyroidectomy is highly successful and to date is not associated with a higher incidence of recurrent HPT.63 Those favoring iPTH as the diagnostic tool believe that all enlarged or hyperplastic glands may not be clinically significant. Harrison has shown that the finding of microscopic hyperplasia in normal-sized glands is of no clinical significance and that the removal of such glands is unnecessary.58 Badder has also proposed that microscopically hyperplastic glands do not have functional significance.57 Questions have been raised regarding the functionality of oncocytic adenomas as well as glands affected by clear-cell hyperplasia.41,64,65 Dawkins has shown that the concentration of PTH per milligram of fresh tissue is 1000 times less in clear-cell hyperplastic glands than in normal glands or in chief-cell adenomas. The clinical significance of this finding is unclear.65
 Please see the Expert Consult website for more discussion of this topic.
Please see the Expert Consult website for more discussion of this topic.Histology
The typical histologic picture for an adenoma or hyperplastic gland is an increased number of chief cells with scattered oncocytes and decreased glandular fat (see Chapter 70, Surgical Pathology of the Parathyroid Glands). Bondeson has noted that some hyperplastic glands, although overall reduced in fat, may have patchy, prominent fat deposits.66 Different histologic categories of adenomas have been identified, including the oncocytic and lipoadenomatous variants. Oncocytic adenomas are uncommon, with one series showing only 6.3% of 160 adenomas being oncocytic.67 Controversy exists regarding whether such oncocytic adenomas are truly functional.36,47,41,64 Lipoadenomas represent a proliferation of parenchymal and stromal elements and are generally associated with clinical hyperparathyroidism.1,41 Histologic variants of four-gland hyperplasia have been described, including clear-cell (Wasserhelle) hyperplasia. Cases of clear-cell hyperplasia were described early in the study of hyperplasia, but subsequent work has shown that this histologic form is rare.1
Molecular Genetics of Primary Hyperparathyroidism
 Please see the Expert Consult website for more discussion of this topic.
Please see the Expert Consult website for more discussion of this topic.
Molecular Genetics of Primary Hyperparathyroidism
Despite new insights into the genetics of hyperparathyroidism, many questions remain unanswered (see also Chapter 70, Surgical Pathology of the Parathyroid Glands). Does the primary genetic defect result in cellular proliferation or a change in calcium/PTH set point, with cellular proliferation occurring secondarily? What is the exact relationship between parathyroid changes and known genetic lesions in MEN 1 (i.e., MENIN) and MEN 2A (i.e., ret oncogene)? Why is there such variable penetrance among the various forms of inherited primary hyperparathyroidism? What are the genetic changes underlying oncogenesis in sporadic disease and parathyroid carcinoma?
Clonality of Sporadic Parathyroid Tumors
 Please see the Expert Consult website for more discussion of this topic.
Please see the Expert Consult website for more discussion of this topic.
Clonality of Sporadic Parathyroid Tumors
Most if not all sporadic parathyroid adenomas are monoclonal lesions arising from a single precursor cell with a selective growth advantage relative to surrounding tissue.68 It is not surprising that parathyroid carcinoma also demonstrates monoclonality.69
Parathyroid Oncogene Rearrangement/Overexpression
 Please see the Expert Consult website for more discussion of this topic.
Please see the Expert Consult website for more discussion of this topic.
Parathyroid Oncogene Rearrangement/Overexpression
Rearrangement and overexpressions of cyclin D1/PRAD1 oncogene result in overexpression of cyclin D1. Cyclin D1 is a target of wvt/β-catenin signaling pathway.70 Aberrations in this pathway have been identified in a variety of parathyroid tumors.71,72 Overexpression of cyclin D1 (PRAD-1), a gene on chromosome 11 that controls the cell cycle, was the first molecularly defined genetic abnormality in sporadic parathyroid tumors. In a small subset of tumors, an inversion within chromosome 11 allows the PTH gene control region to drive inappropriately high expression of cyclin D1. Somatic mutations of the MENIN gene, which when inherited in the gene line causes MEN 1, are found commonly (25%) in sporadic parathyroid adenomas. Widespread somatic mutations in chromosomes 1, 6, 15, and 11, still not defined at the sequence level, suggest that each parathyroid adenoma carries multiple mutations. 68,73–78
Parathyroid Tumor Suppressor Gene Inactivation
 Please see the Expert Consult website for more discussion of this topic.
Please see the Expert Consult website for more discussion of this topic.
Parathyroid Tumor Suppressor Gene Inactivation
Allelic loss (loss of heterozygosity; LOH) is a popular tool for identifying tumor suppressor genes in various forms of neoplasia. LOH at the MEN type 1 locus of chromosome 11q13 is found in ~ 25% to 40% of sporadic parathyroid adenomas, and somatic homozygous mutations of the recently identified MEN 1gene are found in ~ 50% of tumors with LOH at 11q13.79–81 Many genes—including p15INK4d, p16INK4a, RAD51, and RAD54—have been investigated and have been found unlikely to contribute to parathyroid tumorigenesis.82 Klotho has been suggested to be a tumor suppressor gene involved in the regulation of insulin-like growth factor (IGF) signaling in breast, ovarian, and cervical tumor, but its role in parathyroid tumorigenesis is unclear.82
The four-gland hyperplasia form of sporadic primary hyperparathyroidism may initially represent a four-gland polyclonal hyperplastic process from which one or more glands may develop a clonal lesion. The clonal lesion may arise secondary to cumulative risk for mitotic errors or impaired capacity for DNA repair, which characterize the hyperplastic process.68,74,75,83,84 Thus, the difficulty in distinguishing hyperplastic glands from adenomas on a histopathologic basis may in part be due to the fact that many hyperplastic glands are, in fact, adenomas. Arnold has demonstrated tumor monoclonality in at least one gland in 38% of patients with sporadic hyperplasia.85 Thus, hyperplasia is probably not simply a polyclonal response to a systemic growth stimulus. The concept of an underlying germline genetic defect leading to multifocal preneoplastic proliferation as an initial step in tumorigenesis with a second acquired, perhaps inevitable genetic change leading to monoclonal proliferation has been used to explain oncogenesis in retinoblastoma, neurofibromatosis, and medullary carcinoma of the thyroid.50,86–88 Some insight can be gained from investigations of secondary hyperparathyroidism (see Chapter 66, Surgical Management of Secondary and Tertiary Hyperparathyroidism). Some researchers have proposed that secondary hyperparathyroidism involves a shift from initial diffuse polyclonal hyperplasia to nodular clonal formation, presumably because of a secondary acquired genetic change.76 Tominaga found that larger and more nodular tumors were more likely to demonstrate clonality than smaller, anodular glands.76,89 Others have disputed this finding.85 Gagne has found the recurrence rate to be higher after parathyroidectomy in uremic patients with nodular versus diffuse hyperplasia.90 Cytometric DNA analysis suggests greater growth potential in parathyroid cells within nodules of nodular hyperplasia as opposed to diffuse hyperplasia.89 These data suggest that clonal events represent an evolution of multiglandular disease and are associated with advanced clinical presentations. The PRAD-1/cyclin D-1 overexpression and chromosome 11q13 allelic loss, which characterize some clonal proliferation in sporadic adenoma of primary hyperparathyroidism, appear not to be important in the clonal formation and progression of nodular hyperplasia in secondary hyperparathyroidism.76,85 Other acquired genetic factors may be involved and may relate to the calcium-sensing receptor gene or a vitamin D receptor mechanism.
Parathyroid Surgical Anatomy
Normative Parathyroid Parameters
Parathyroid Number
The characteristics and location of the parathyroid glands are discussed in detail later in this chapter. Here we will simply review parathyroid number and weight. Humans typically have four parathyroid glands. Several autopsy studies of normal individuals without hyperparathyroidism have reported that 3% to 6% have fewer than four parathyroid glands. Wang has suggested these data derive from inadequate dissection and that the minimum number of parathyroids should generally be regarded as four.91–95 These same studies show that more than four glands occur in 2.5% to 6.7% of cases.91–95 Akerstrom noted that some of the supernumerary glands are very small rudimentary glands or split glands, typically within the thymus or in the fat adjacent to the normal parathyroid gland. Such small rudimentary glands may be present in up to 13% of normal individuals. True supernumerary glands (defined as greater than 5 mg and located at some distance from the normal parathyroid gland) occur in only 5% of cases.94 Such supernumerary glands in patients with primary hyperparathyroidism are most often found within the thymus. In studies of patients with primary hyperparathyroidism, a fifth parathyroid gland is found in from 0.6% to 0.7% of patients.93,96,97 Supernumerary glands also occur in secondary hyperparathyroidism. Edis and Levitt found 10% of persistent hyperparathyroidism after surgery in patients with secondary hyperparathyroidism resulting from supernumerary glands.98
Parathyroid Weight
The normal weight of the parathyroid gland increases until the third to fifth decade of life. Parathyroid gland weights are lower in chronic, nonrenal disease states. The lower glands weigh slightly more than the upper glands99 The upper limit of normal parathyroid gland weight ranges from 38 to 59 mg.99–101 Bonger has noted that there is overlap between normal and abnormal gland weights and has found hyperplastic glands weighing only 60 mg.102 The clinical significance of such small, but hyperplastic, glands is discussed earlier.
Parathyroid Gland Characteristics: the “Gliding Sign”
Understanding parathyroid gland embryology and intraoperative attention to detail facilitates the identification of normal and abnormal parathyroids. Parathyroid glands can be seen as distinct from surrounding neck tissues through recognition of a number of different characteristics (see Table 22-4). The color of the parathyroid gland is typically light brown to reddish tan and is often described as mahogany. This color relates to the fat content, vascularity, and percent of oxyphil cells.103 In obese persons, the fat content of parathyroid glands is higher. As a result, they assume a more yellowish color, which is only slightly different from surrounding fat.41 In infants younger than 3 months of age, the normal parathyroid gland looks grayish and semitranslucent. In children the normal parathyroid gland, which has scant fat, is darker (a pinkish brown) compared with that of adults. The normal adult parathyroid gland has greater fat content than that of a child and is more yellow. With increasing adult age, the parathyroid gland darkens.104 In adults, hypercellular glands with less fat are browner. Large glands associated with secondary hyperparathyroidism can be grayish. A gland involved by parathyroid carcinoma can appear white, hard, and irregular, with a surrounding fibrotic change associated with local invasion.102,105 Fat, in contrast, is bright yellow. The thyroid is firmer and mottled reddish brown, whereas lymph nodes vary from gray to tan to red and are generally firmer than parathyroid glands. Parathyroid glands also have a distinct hilar vessel (a “vascular strip”) that can be seen if surrounding fat does not obscure its hilum. The parathyroid gland has a discrete, encapsulated, smooth surface as opposed to the more lobular surface of the thyroid and the more mottled, pitted surface of lymph nodes. The parathyroid gland is softer to palpation than thyroid or nodal tissue. Parathyroid shape is also unique and most often leaflike or beanlike.93 The dissection of a small nodule on the surface of the thyroid gland mistaken for a parathyroid gland results in hemorrhage, whereas the dissection of a parathyroid gland closely related to the thyroid capsule does not result in significant hemorrhage when performed with meticulous loupe-assisted dissection. The most important distinguishing feature is that the parathyroid, being a discrete, encapsulated organ, has a characteristic motion when surrounding fat is gently manipulated. The discrete edge of the parathyroid gland can be seen as this fat is dissected. As the surrounding fat is manipulated, the parathyroid gland demonstrates a discrete gliding motion within the fat, like that of a rowboat riding on a wave of water. As Wang has written of the superior parathyroid gland, “The absence of capsular fixation permits an impression of mobility uniquely characteristic of the parathyroid.”93 This “gliding sign” is a tremendously helpful clue in parathyroid recognition during central compartment surgery.



