1

Pharmacology of
Botulinum Neurotoxins
Botulinum neurotoxin (BoNT) has been approved as a safe and effective therapeutic option for several clinical conditions. The common basis underlying all of the conditions amenable to treatment with BoNT is overactivity of neurotransmitter or neuropeptide release. The clinical efficacy of BoNT is based on the localized and highly specific, but reversible, disruption of the exocytotic mechanism within neurons, resulting in temporary inhibition of the release of neurotransmitters after injection into specific muscles or other tissues.
Botulinum neurotoxins are synthesized by a variety of clostridial species, most notably Clostridium botulinum, but also including C. baratii and C. butyricum.1 These bacteria synthesize several immunologically distinct neurotoxins classified as serotypes A through G. The biologic activity of the neurotoxin is contained within a 150-kd protein commonly referred to as the core neurotoxin. In nature, a single 150-kd neurotoxin is incorporated into a molecular complex that varies in size based on the number of associated nontoxic proteins, which are classified as hemagglutinins or nonhemagglutinins.2 The largest neurotoxin complex is 900 kd, formed only by the A serotype. Serotypes A, B, C1, and hemagglutinin-positive D are in the form of 500-kd and 300-kd complexes, whereas serotypes E, F, and hemagglutinin-negative D form only 300-kd complexes.3 The neurotoxin complex is known to stabilize and protect the core neurotoxin against thermal and pH stress in addition to shielding the neurotoxin from enzymatic degradation.4–6
The core neurotoxin is synthesized as a 150-kd single-chain protein that must be cleaved or “nicked” by proteases to exert its activity.7 Cleavage produces a di-chain molecule consisting of a 100-kd heavy chain and a 50-kd light chain that are held together by a disulfide bond.
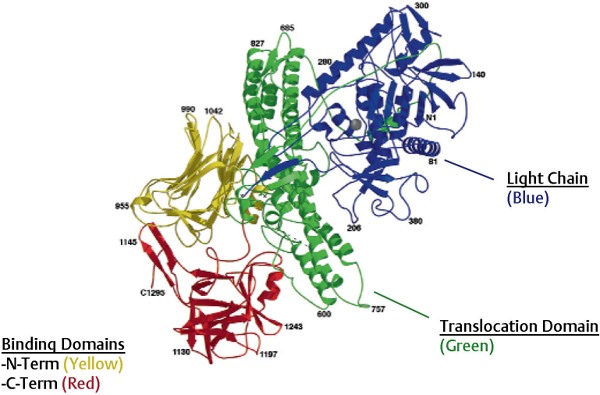
Fig. 1.1 Botulinum neurotoxin (BoNT) molecular structure. (From Lacy DB, Tepp W, Cohen AC, et al. Crystal structure of botulinum neurotoxin type A and implication for toxicity. Nat Struct Biol 1998;5:898–902. Reprinted by permission.)
The activated di-chain molecule consists of three functional domains, each playing a defined role in the mechanism of action of the neurotoxin: the C-terminal portion of the heavy chain contains the binding domain, responsible for docking the neurotoxin to the neuronal surface; the N-terminal portion of the heavy chain contains the translocation domain, whereas the light chain contains the catalytic domain responsible for cleavage of the intracellular target of BoNT (Fig. 1.1). The tertiary structure of the di-chain protein is conserved through all clostridial neurotoxins; however, there is considerable heterogeneity between the protein sequences of the different serotypes (reportedly up to 70%), accounting for their different neuronal affinities, antigenicity, and intracellular targets.8
In the treatment of skeletal muscle overactivity BoNTs inhibit acetylcholine release at the neuromuscular junction, thereby reducing excessive muscle contractions. This inhibition of calcium-dependant vesicular neurotransmitter release occurs via a multiple-step process.
In the first step, the neurotoxin must dissociate from its protective protein shield to allow binding of the free neurotoxin to the neuronal surface. The kinetics of dissociation in vivo are currently unknown, but the stability of the neurotoxin complex is known to be affected by basic pH and increasing ionic strength of the solute.9,10
The binding of the free BoNT to the neuron is accomplished through the interaction of the heavy chain of the 150-kd neurotoxin with neuronal receptors that are located predominantly, but not exclusively, on cholinergic nerve terminals. For BoNT/A (the most commonly used in clinical practice), a two-receptor mechanism has been proposed: the neurotoxin first interacts with a cell-surface resident ganglioside that serves to retain the neurotoxin close to the plasma membrane, facilitating the binding of the neurotoxin with its cognate protein receptor. The protein receptor for type A, E, and F neurotoxins has been identified as SV2, a constituent protein of the synaptic vesicle.11–14 The neurotoxin is only able to bind to SV2 when the protein is exposed on the cell surface during neurotransmitter release. Thus, the neurotoxin preferentially enters neurons that are actively secreting neurotransmitter or neuropeptides.11 The receptor molecules for serotypes A and B are distinct, with the protein receptor for types B and G identified as another synaptic vesicle protein known as synaptotagmin.13 Other serotypes may also bind to unique sites, but they have not been characterized at this time.
After binding, BoNTs are internalized into the neuron via receptor-mediated endocytosis.15 The neurotoxin undergoes a conformational change inside the acidified endocytotic vesicles, allowing the translocation domain to interact with the membrane of the endocytotic vesicle, providing a portal to enable the light chain to traverse the wall of the endosome.16 The low pH in the endocytotic vesicle also reduces the disulfide bond, which releases the light chain from the heavy chain, freeing it to enter the neuronal cytosol.17 Once inside the cytosol, the light chain, which contains a zinc-dependent endopeptidase, disrupts one or more of the SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins necessary for vesicle docking and fusion, thereby reducing exocytotic neurotransmitter release18 (Fig. 1.2). Each serotype cleaves a specific peptide bond on one or more of these proteins. Type A neurotoxin cleaves the membrane-associated SNARE protein SNAP-25, whereas type B neurotoxin cleaves the vesicular-associated protein VAMP (synaptobrevin). It has been recently proposed that modulation of the interstitial zinc concentration might alter the clinical efficacy of BoNT activity; however, this concept remains unexplored at this time.
The relative duration of inhibition of neurotransmitter release varies among serotypes, presumably based on the half-life of the light-chain19 and the time it has taken for the neuron to restore intact SNARE proteins (Fig. 1.3). In a preclinical model, the duration of effect was the longest for type A, followed by types C1, B, F, and E.20 Following injection in human muscles, serotypes A and C1 appear to have the longest duration of effect.21
The recovery of neuronal activity following BoNT–induced denervation has been examined in several preclinical models.22–24 Early studies showed that axonal sprouts developed from the affected nerves, likely in response to growth factor secretion from the denervated muscle. These sprouts are active and produce temporary reinnervation during the early recovery phase. The exact duration of the pharmacologic action of BoNT in neurons is not known; however, during the later phase of recovery, vesicular release has been found to return at the original terminal, a process that is accompanied by retraction of the neuronal sprouts. This suggests a process of sprouting that is associated with reestablishment of activity over time in the originally affected nerve terminal.
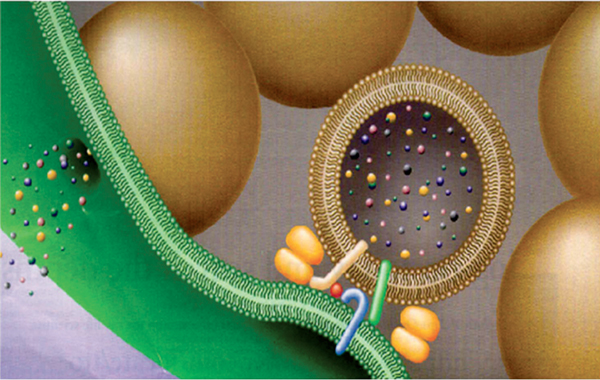
Fig. 1.2 Inhibition of exocytosis. (From Martin TF, Stages of regulated exocytosis. Trends Cell Biol. 1997 Jul;7(7):271–6.)
The inhibition of neurotransmitter release by BoNTs is reversible, which has both benefits and limitations in clinical use. The benefits include the flexibility to inject a given muscle based on its activity level, which may fluctuate over time, the avoidance of permanent interventions such as surgery, and the resolution of any unintended effects of BoNT treatment. The primary limitation associated with the reversibility of the effects of BoNT is the need for repeated injections; however, repeat treatment can result in sustained efficacy. As most conditions treated with BoNT are chronic in nature, a longer duration of effect is a desirable property and was the rationale for the development of type A over other serotypes of BoNT for clinical use.
In addition to the inhibition of acetylcholine release from alpha motor neurons, BoNT/A also inhibits neurotransmitter release from gamma motor neurons innervating the intrafusal fibers of the muscle spindle.25–27
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


